
下载掌阅APP,畅读海量书库
立即打开



微生态(micro-ecology)是存在于植物或动物体内的包括共生微生物和病原微生物的共生生态群落。人体微生态是近年发现的具有重要作用的“新器官”,其在维持人体健康和疾病的发生发展过程中发挥着不可或缺的作用。微生物群(microbiota)主要包括细菌、古菌、原生动物、真菌和病毒等具体微生物种类;微生物组(microbiome)是指一个特定环境或者生态系统中全部微生物及其遗传信息,包括其细胞群体和数量、全部遗传物质(基因组)。它界定了涵盖微生物群及其全部遗传与生理功能,其内涵包括了微生物与环境和宿主的相互作用。微生物组学(microbiomics)是以微生物组为对象,研究其结构与功能、内部群体间的相互关系和作用机制,研究其与环境或宿主的相互关系,并最终能够调控微生物群体生长、代谢等,为人类健康和社会可持续发展服务的学科。在对人体微生物群的描述中常用到的指数包括α多样性和β多样性。α多样性主要关注局域均匀生境下的物种数量,因此也被称为生境内的多样性(within-habitat diversity)。β多样性指沿环境梯度不同生境群落之间物种组成的相异性或物种沿环境梯度的更替速率,也被称为生境间的多样性(between-habitat diversity)。不同群落或某环境梯度上不同点之间的共有种越少,β多样性越大。精确测定β多样性具有重要意义,这是因为:①它可以指示物种被生境隔离的程度;②β多样性的测定值可以用来比较不同地段的生境多样性;③β多样性与α多样性一起构成了总体多样性或一定地段的生物异质性。
在跨学科研究的帮助下,临床医师尝试了解微生物群在各个器官中所扮演的角色,然而这些微生物群与宿主之间所产生的相互作用以及这种相互作用在健康和疾病之间的关系是非常复杂的。对于研究者而言,要想在短时间内探索这种共生关系绝不可能,因为随着研究深入,这一共生关系愈发复杂。自微生物进入学者的研究中以来,国内外主要聚焦于探索肠道微生物,对于泌尿道微生物的研究也是近些年才开展。目前研究人员已经使用高通量测序和增强尿培养(expanded quantitative urine culture,EQUC)技术发现并确认人体尿路中的微生物,由此“健康人泌尿道是无菌状态”这一教条理论被打破。
随着对尿路细菌微生态的深入研究,泌尿道微生物研究领域的权威学者Alan J Wolfe教授在2018年首次提出概括性术语“urobiome”一词来表达尿液中的微生物群及微生物组。前期的临床研究已经发现尿路微生物与泌尿系统疾病之间存在关联,然而同人体其他部位的微生物组一样,在尿路微生物组研究中仍需要深入探索其与泌尿系统疾病的潜在机制关系。
人体的尿路微生物群主要是由细菌、古生菌、病毒和真菌组成,在性别不同的人群中尿路微生物群不尽相同,并且随着年龄变化微生物群可能也会发生改变。
对于尿路微生物群,我们尚只看到零光片羽。本章将从正常尿路微生态和疾病状态下尿路细菌微生态两个方面进行阐述,并对尿路细菌微生物与泌尿系统疾病的临床表现、诊断、治疗原则等临床问题进行讨论。
(吴芃 编 郑波 审)
近些年来,随着对人体微生态的研究逐渐加深,人们才开始接受在人体的大多数部位并非无菌状态,并且这些细菌主要为非致病菌。然而在医学研究中,传统思想关注到的主要为致病菌,因此人们对于人体自然居住或共生菌的认知少之甚少。实际上,在健康状态下,人体内也存在着各种微生物,如细菌、真菌、病毒和原生动物,并且人体内所包含的微生物细胞大约是人体细胞的10倍。
人类微生物组计划(human microbiome project,HMP)最初于2008年启动,目的是对人类微生物组进行全面描述,并分析其在人类健康和疾病中的作用。2012年,HMP对242名健康人进行了调查。此项工作为研究人员提供了一个初步用来描述健康和疾病人群中微生物群落的框架。尽管在对如何定义“有益”微生物群这一结论时仍存在着许多争议,但就目前研究进展而言可以得出如下比较重要的结果:首先,微生物群落的特征通常不只依赖其中某一种微生物的属性,而以某一组微生物的属性为特征;其次,尽管不同群体的微生物群落各有其特点,但“有益”微生物所发挥的作用往往是相似的。
处理尿液通常采用标准尿培养的方法,这种方法主要针对尿液中快速生长的需氧微生物,而对检测尿液中厌氧及生长缓慢的微生物有很大局限性,导致业内长期以来认为在健康人群的膀胱和尿液中是无菌的。因此,在HMP成立早期,并没有涉及对尿液微生物群的研究,使得对尿路微生物组的研究落后于人体其他微生物组的研究数年。随着分子生物学技术和培养方法的进步,如16s核糖体核糖核酸(ribosomal RNA,rRNA)测序和增强尿培养(EQUC)技术等的出现,使人们对尿路微生物的认知变得更加全面。EQUC技术结合了多种不同的培养基、需氧和厌氧条件以及不同的生长温度,能够分离培养出尿液中80%的细菌,而在标准尿培养的结果中,大多数没有细菌生长(图1-2-2-1)。
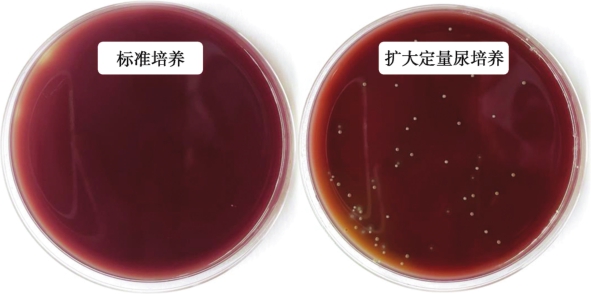
图1-2-2-1 EQUC技术的优势
虽然我们对于尿路微生物的认识还处于初级阶段,且目前仍有许多未知需要探索,但这些开创性的研究改变了临床上对于“膀胱中是无菌环境”“泌尿道症状是由于某单一病原体感染导致”的刻板认知。研究者们结合在尿路微生物群和微生物组的新发现,将有助于临床治疗和对疾病发病机制的理解。那么这一过程中,最重要的一步就是了解正常尿路微生态的特征,以确定群落中微生物组成与临床疾病之间的联系。尽管缺乏对健康人群中微生物群落的全面认识,但能确定的是,微生物失调(如有益菌的缺失)与多种临床疾病相关,如炎症性肠病、多发性硬化症、糖尿病、过敏、哮喘、自闭症和肿瘤。同样,利用微生物失调这一概念解释泌尿系统疾病的发生,是非常有意义的。这使得临床医师意识到,某些泌尿道疾病可能是由微生物群落失调引起的,而不是单纯的病原体入侵,这也为预防、诊断和治疗疾病提供了新方法。
目前检测、培养尿液微生物的方法逐步趋于成熟,但是样本间存在性别、数量以及采样方式的差异,也会导致测序及培养结果的不同,这一点在对尿路微生物的研究中是非常重要的。现阶段,在对尿路微生物的报道中,在属水平上,乳杆菌属和链球菌属是最常被报道的菌,并且在属水平中皆都为乳酸菌,其在包括泌尿道在内的体内组织中发挥着抵御病原体的保护作用。相比而言,如伯克霍尔德菌属、克雷伯菌属、罗河杆菌属、韦荣球菌属则在泌尿道中检出率较低。
起初,女性尿路微生物检测是十分困难的,主要有两个原因:第一,在获取膀胱中尿液时必须防止其被阴道中的细菌所污染;第二,检测到的DNA必须证明其来源为活菌。为了克服第一个难题,美国芝加哥洛约拉大学的研究者采取耻骨上穿刺法(suprapubic aspiration,SPA)从膀胱中直接获取尿液样本,此方法收集的尿液可避免被外阴及阴道污染的可能。随后,使用16s rRNA测序的方法,将SPA样本中的细菌DNA与经尿道导尿管(transurethral catheter,TUC)获得的样本中的细菌DNA进行比较。为了判断获得的样本是否被阴道所污染,将TUC样本与中段尿样本和阴道拭子进行比较。结果发现,SPA和TUC尿液菌群具有相似的特征,但与中段尿和阴道拭子则不同,因此可以得出膀胱中含有细菌DNA的结论。那么,另外一个问题随之而来,如何判断检测到尿液中的细菌DNA来自活细菌,而不是死亡的细胞DNA或尿液中游离的DNA呢?洛约拉大学的研究者在先前的工作中已经对膀胱中能检测到的细菌利用16s rRNA测序技术进行了分类鉴定。虽然大多数检测到的细菌是可培养的,但由于标准尿培养(将1μL尿液在血平板和麦康凯琼脂培养基培养24h)的条件单一且时间较短,导致大部分菌群无法生长,因此一种增强尿培养(EQUC)技术被推行。与标准尿培养不同,EQUC将更多的尿液(100μL)在多种培养基上培养48h,并在不同的大气培养环境(如高浓度CO 2 、缺氧)下培养。与测序结果相对应,大部分检测到的微生物可以通过此方法培养出。
群体分型(分层/层化,stratification)是一种有助于更好地理解人类身心健康等复杂生物问题的有效方法。2011年,MetaHIT团队将这种方法应用到肠道微生物中,提出“肠型”的概念,由此可以将不同的群落组成定义为肠型,这对描述肠道微生物群落结构非常有效。而随着对尿路微生物的探索,同样可以将此方法进行合理的使用,越来越多的研究者逐渐认可“尿型”这一概念。这种方法有助于对某个群落进行描述,并可能用于指导临床实践。当然,在每个群落中也可以使用生态参数进行描述,如多样性、稳定性或恢复力。在对无泌尿系统疾病的成年女性中导尿样本的研究发现,不同尿型的多种尿路微生物群落的状态和结构与尿路维持健康状态有关。在健康人群尿型中,最常见的主要是由乳酸菌属构成,这种属同样寄生于阴道内。虽然尚有证据支持女性泌尿道与生殖道内的乳酸菌属可能为同一来源,但此结论仍有待考证。事实上,乳酸菌属被认为在身体的许多部位发挥着保护作用。其他常见的尿型主要由加德纳菌属、链球菌属、葡萄球菌属、棒状杆菌属和大肠埃希菌属构成。随着年龄变化,尿型也会发生变化。比如以加德纳菌属为主的尿型在绝经前女性中更常见,而以大肠埃希菌为主的尿型在绝经后女性中更常见。即使在短期内,尿型会有一定的波动性,但长期观察发现尿型仍是处于稳定状态,其往往会在以乳酸菌属为主的群落和以加德纳菌属为主的群落中相互转换。导致这种短期内的波动与两种因素有关,一种是内在因素(月经周期),另一种是外在因素(通过阴道进行性交)。然而尿路微生态似乎有某种弹性,能够在数天内恢复到正常状态。
成年女性的尿路微生态与发生泌尿道感染(UTI)的易感性有关。尤其在进行盆腔手术导尿或其他经尿路操作等高危时期,尿路微生态更容易受到影响。性交、妊娠后女性的UTI患病率会有所升高,这也有可能与尿路微生态有关。因此,对尿路微生态的特征进行风险分层,可以有效地进行具有针对性措施以预防和改善临床症状。除此之外,成年女性的尿路微生物与常见的尿失禁有关,主要包括急迫性尿失禁(urgent urinary incontinence,UUI)和压力性尿失禁(stress urinary incontinence,SUI)。在一项对接受口服索利那新治疗UUI的女性中,发现药物疗效似乎与治疗前的尿路微生物有关。而其中在对药物具有明显疗效的女性中发现,症状获得改善的尿路微生物具有与正常尿路微生物相似的特征。这些尿路微生物群中物种很少,并且通常是以乳酸菌为优势菌种。相比之下,治疗无效的女性具有更复杂的微生物群并且没有优势菌。这也为尿路微生物群作为抗胆碱能药物(索利那新)治疗反应的预测因子提供了可能。此外,药物的治疗反应同样与尿路微生态的恢复力和稳定性有关,目前仍需要更多的研究来佐证其与药物治疗反应之间的关系,由此帮助临床医师提供和指导个性化治疗,改善患者预后。
早期研究表明,男性排尿获取的尿液微生物组与远端尿道中的微生物组相似,此外,在有性传播感染和非性传播感染的男性尿液微生物组是不同的,并且尿道微生物组可能受到性活动的影响。显然,在研究尿路微生物时,如何避免样本的污染以及确保样本的代表性无疑是至关重要的。在男性中,晨尿样本所检测到的微生物群与远端尿道拭子检测到的微生物群落则非常相似。种种迹象表明,对尿液进行分析检测时,尿液所携带的近端尿道中的微生物会被远端尿道中含量较高的微生物所掩盖。在一项比较使用TUC采集和中段尿采集的尿液样本中发现,在中段尿样本中,通过EQUC技术检测样本的细菌检出率为96%,16s rRNA测序技术检出率为80%,总检出率为98%。而在TUC样本中,相应的细菌检出率分别为29%、27%、39%。
最新的研究证实,男性尿路微生物与下尿路症状(lower urinary tract symptoms,LUTS)有关。良性前列腺增生(benign prostatic hyperplasia,BPH)是老年男性常见疾病,常伴发尿频、尿急等泌尿系统症状。通过将BPH患者尿液与正常男性尿液进行分析,结果表明其尿路微生物间存在明显差异。并且,在对BPH患者进行国际前列腺症状评分(international prostate symptom score,IPSS)中发现,分数的增加与男性膀胱内微生物存在相关性,并且随着尿液中微生物的检测率增加,IPSS评分的严重程度也随之增加。然而,由于人们对这些给人类带来痛苦的疾病了解较少,目前也没有有效的治疗手段,因此推进对尿液微生物组的研究,为患有慢性前列腺炎/慢性骨盆疼痛综合征的男性带来了新的希望。
而这种微生物之间的差异不只出现于BPH患者中,在诊断为前列腺癌患者的尿液微生物群也存在着显著差异。有趣的是,前列腺癌组患者的精液和前列腺液中大肠埃希菌数量较高,而尿液中大肠埃希菌数量则较低。此外,前列腺癌患者精液中肠球菌数量较高,而尿液和前列腺分泌物样本中肠球菌数量无显著差异。若将微生物不视为前列腺癌的致病因素,而将其作为前列腺癌患者诊断或对治疗反应的检测指标,是否更具有临床意义,值得研究者们深入讨论。
(吴芃 编 郑波 审)
随着现代培养和测序技术的发展,人类尿路细菌微生态逐步进入人们的视野,越来越被认为是影响人类健康和疾病的重要因素。现有的技术已经能够检测整个泌尿系统的微生物,在迄今为止发表的所有研究中,乳杆菌( Lactobacillus )和链球菌( Streptococcus )是在尿路细菌微生态中最常被报道的菌属。越来越多的研究表明,尿失禁、膀胱癌(bladder cancer,BCa)、前列腺癌、肾癌、肾结石、神经源性膀胱功能障碍(neuropathic bladder dysfunction,NBD)、间质性膀胱炎(interstitial cystitis,IC)、慢性前列腺炎/慢性骨盆疼痛综合征(chronic prostatitis/chronic pelvic pain syndrome,CP/CPPS)等泌尿系统疾病患者的尿路细菌微生态存在着不易感知的变化。
关于微生物组的影响及其在健康和疾病中的功能作用有了重大进展。鉴于共生微生物组对人类健康和疾病众多状态的影响,目前已成为研究热点。现有证据表明,结直肠腺瘤、腺癌以及胃癌和胆道癌与微生物存在关联,特别是细菌微生态。细菌可以通过干扰β-catenin信号通路等机制促进癌症的发生,也可以通过代谢途径及产生致癌化学物质(如亚硝胺和乙醛)来调节癌症发生风险。对健康和疾病状态的连续性研究进一步表明,尿路细菌微生态能够影响泌尿系统的状况。相关细菌微生态的确切性质和作用仍在研究中,但它们的潜在参与现已变得愈发明显。
膀胱癌是我国泌尿外科临床上最常见的恶性肿瘤之一。世界范围内,其发病率居恶性肿瘤的第9位。膀胱癌的发生是复杂、多因素、多步骤的病理变化过程,既受内部遗传因素的影响,又受外在环境因素,包括慢性感染[如细菌、血吸虫、人乳头状瘤病毒(human papilloma virus,HPV)感染等]的影响。现有研究表明,与膀胱鳞状细胞癌相关的感染危险因素,如血吸虫感染,可诱导内源性N-亚硝胺和氧自由基的合成,从而诱发膀胱鳞状细胞癌的发生。尿路细菌微生态可能通过调节内源性抗肿瘤免疫反应参与膀胱癌的各个发展阶段。新近研究表明,膀胱癌患者的细菌丰度较健康人群明显升高。根据欧洲癌症研究和治疗组织(European Organzation for Research and Treatment of Cancer,EORTC)评分系统,复发或进展风险高的非肌层浸润性膀胱癌(non-muscleinvasive bladder cancer,NMIBC)患者的尿液中也存在更丰富的细菌。例如,链球菌在膀胱尿路上皮癌患者的尿液中富集,但健康人群中尿液的链球菌丰度都接近零,其次在链球菌丰度较低的膀胱尿路上皮癌样本中,发现假单胞菌属和厌氧菌属的丰度最高。在门水平上,丰度最高的门是厚壁菌门( Firmicutes ),其次是放线菌门( Actinobacteria )、拟杆菌门( Bacteroidetes )和变形菌门( Proteobacteria )。另有报道指出,鲍曼不动杆菌的毒力因子具有包括上皮细胞的侵袭、磷脂的降解和生物膜形成的作用,这些都有助于逃脱宿主的免疫反应,以及厌氧菌可诱导炎症和细胞外基质(extracellular matrix,ECM)重构,可能在膀胱癌发病、进展和复发中发挥关键作用。现有的各项研究结果表明,较高的细菌丰度可能是NMIBC复发和进展风险较高的潜在指标。
除了研究尿路细菌微生态和肿瘤发生之间的联系外,对治疗的影响也在研究中。随着潜在联系被日益重视,通过控制细菌微生态来降低风险或预防疾病复发的可能性变得愈发有吸引力。早在1992年就有学者提出口服抗菌药物对膀胱肿瘤电切术(transurethral bladder tumer resection,TURBT)后复发有预防作用。此外,降低膀胱癌的发生风险,其主要是通过刺激中性粒细胞分泌细胞因子,诱导树突状细胞成熟和产生抗原特异性的细胞毒性T细胞对抗癌细胞。亦有研究表明,使用选定的细菌诱导抗原特异性细胞毒性可能成为对抗癌细胞的新选择。
运用卡介苗(bacillus calmette guerin,BCG)来预防和降低NMIBC患者TURBT术后复发的风险已在临床中广泛应用。BCG中使用的减毒结核分枝杆菌与膀胱癌之间已建立起有益的联系。BCG被认为是通过纤维连接蛋白来刺激免疫反应,从而进入膀胱细胞。现有证据证实,BCG接种到膀胱后,引发细菌炎症反应,诱导抗肿瘤免疫效应。某些特定的共生细菌黏附在纤维连接蛋白上,从而占据卡介苗发挥效用的结合位点,这将降低BCG的效用,并可能降低清除肿瘤细胞所需的强细胞毒性反应。
在现有的诸多研究中,乳酸菌是被经常提及的一种益生菌。研究表明,乳酸菌可以诱导抗增殖和细胞毒性作用,降低膀胱癌的发病率和复发风险。灌注益生菌可能是直接影响膀胱细菌微生态的一种潜在策略,但也可能通过间接机制进行调控。
目前,尿路细菌微生态已成为膀胱癌研究的一个新焦点,其与免疫功能之间的联系尚未被广泛报道,尿路微生物组正成为影响膀胱癌发生、发展和治疗反应的重要因素。
早在2017年就有学者提出,在前列腺癌患者中,葡萄球菌属在肿瘤及瘤周组织中富集度更高。Feng等人首次使用宏基因组和转录组学分析来鉴定65例中国前列腺癌患者的肿瘤和邻近良性组织的冰冻标本中的微生物群,共鉴定了超过40种细菌属,其中假单胞菌属、大肠埃希菌属、肠杆菌属、不动杆菌属和丙酸杆菌属的数量最为丰富,但是不同组织类型之间的微生物群特征没有显著差异。只有生殖道支原体与前列腺癌独立相关。运用免疫荧光和RNA测序在标本中发现,痤疮丙酸杆菌是尿路细菌微生态中特殊的一组,亦与前列腺癌的发展有关。
最近的一项研究表明,前列腺癌患者的前列腺液、尿液和精液中的微生物群落存在显著差异,应用前列腺液变性梯度凝胶电泳测序和系统发育分析结果显示,丰度最高的门分别是拟杆菌门、变形菌门、厚壁菌门、放线菌门。前列腺癌患者的精液和前列腺液样本中大肠埃希菌的数量也较高,而尿液样本中数量较低;精液样本中肠球菌数量较高,而尿液和前列腺液样本中无显著差异。
虽然已有相关研究证实前列腺癌患者的肿瘤组织和瘤旁组织在细菌微生态方面存在显著差异,但微生物组在前列腺癌病理生物学中的可能作用仍不明确,需要进一步的研究来阐明两者之间的关系。
肾细胞癌(renal cell carcinoma,RCC)是最常见的癌症类型之一。发病率逐年上升,全球每年确诊的新病例超过40万,死亡人数约为17.5万。虽然在过去的数十年里,靶向药物和免疫检查点抑制剂的发展为RCC患者提供了巨大的临床益处,但为了进一步降低RCC的死亡率,更好地了解RCC的发病机制仍是必要的。近年来,越来越多的证据表明,尿路细菌微生态可能在RCC的发生发展中发挥重要作用。随着二代测序技术的应用,越来越多的人认识到微生物群对癌症的影响。最近在尿路中发现的一种独特的微生物群,揭示了微生物群与肾脏之间的直接相互作用程度尚不被充分认识的可能性,尽管这种相互作用与肾细胞癌(RCC)发病机制的关系尚未被研究。随着在物种水平上鉴定特定细菌的技术出现,这可能会促进尿液微生物组与RCC发病率的进一步研究。
国内有学者通过16s rRNA基因测序,对RCC组织及其邻近正常组织中的微生物群进行了分析,以阐明微生物群在RCC发病和发展中可能的作用。在这项研究中,分析了24例RCC患者的肿瘤组织和配对正常组织中的微生物群。在门、纲、目、科、属水平上的微生物分类概况概述了微生物区系的变化,在门水平,相对丰度最高的门分别是变形菌门、厚壁菌门、拟杆菌门和放线菌门;纲水平上相对丰度最高的是α-变形杆菌纲、β-变形菌纲、γ-变形菌纲、杆菌纲;目水平上相对丰度最高的是根瘤菌目、伯克霍尔德菌目、假单胞菌目;科水平上相对丰度最高的是布鲁氏菌科、丛毛单胞菌科、莫拉菌科、 Chitinophagaceae ;属水平上相对丰度最高的是腐生螺旋体属( Saprospirales )、苍白杆菌属、不动杆菌属、沉淀物杆状菌属( Sediminibacterium )、贪铜菌属( Cupriavidus )。
基于16S rRNA扩增子测序结果预测微生物群落功能(phylogenetic investigation of communities by reconstruction of unobserved states,PICRUSt)分析确定的微生物群落预测功能谱,RCC组织与正常组织之间的9条京都基因和基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路存在显著差异。其中,RCC组织中富集3条通路(膜运输、转录、细胞生长与死亡),而相邻正常组织中富集6条通路(细胞运动、信号转导、代谢、辅因子与维生素代谢、能量代谢、内分泌系统)。
亦有学者收集了10例6个月内无尿路感染史的肾癌患者,行腹腔镜肾切除术后获得组织切片,结果在所有肾脏样本中均发现了大量的微生物,且在良性和恶性肾组织之间存在显著差异。这表明健康肾组织和肾细胞癌组织具有特定的微生物组,从而为肾脏生理学和肿瘤发病机制开辟了新的视角。
现有研究多为回顾性研究,较难确定微生物群变化与RCC之间的因果关系,因此需要更多的大规模前瞻性研究来进一步阐明尿路细菌微生态与肾癌之间的因果关系。
肾结石是世界范围内一种常见的泌尿系统疾病。不同地区的患病率为5%~20%,10年随访复发率为50%,部分患者可发展为慢性肾病或终末期肾病。虽然肾结石发生率高,并发症严重,但其形成的病理生理机制尚不完全明确。肠道微生物组在尿液草酸排泄和肾结石形成中的作用一直是研究的热点,研究表明可能与形成肾结石的病理生理过程相关,但肾结石与尿路细菌微生态的关系仍在探索中。产甲酸草酸杆菌( Oxalobacter formigenes )是一种草酸降解菌,在过去的研究中被证实与尿结石的产生呈负相关。已有相关文献报道,尿路微生物组比肠道微生物组更能引起尿结石。
细菌很早就被认为是导致磷酸镁铵类结石的原因,然而其在更常见的草酸钙(CaOx)和磷酸钙(CaPhos)结石形成中的作用尚未被广泛研究。75%的肾结石主要由草酸钙组成,尿草酸盐被认为是一个关键的危险因素。在临床中,无论结石成分如何,泌尿系统结石患者通常伴有泌尿系统感染。一些研究结果表明,尿结石可能与细菌有关。这些细菌可能来自尿液或结石本身。基于16s rRNA基因测序和增强培养(EQUC)技术,有研究者为了确定结石中细菌富集程度是否高于尿液,鉴定了肾结石患者的尿液和结石微生物群。结果表明,与膀胱尿液相比,对结石样本使用EQUC后可以分离出表皮葡萄球菌、阴沟肠杆菌、大肠埃希菌和加氏乳杆菌等细菌,而上尿路的尿液与膀胱尿液相比,菌群组成无明显差异。研究者亦比较了肾结石患者的膀胱尿液和健康人群的膀胱尿液在不同分类水平上的微生物群的相对丰度。在门水平上,拟杆菌门( Bacteroidetes )、变形菌门( Proteobacteria )和厚壁菌门( Firmicutes )的平均丰度差异具有统计学意义。在其他分类学水平上,两类人群的膀胱尿液也存在着显著的丰度差异,在属水平上最具代表性的是肾结石患者中的不动杆菌属和健康人群中的普氏菌属。肾结石患者的膀胱尿液和肾盂尿液中的粪杆菌属和乳酸杆菌属的相对丰度明显低于健康人群。与此同时,与健康人群的膀胱尿液相比,肾结石患者的膀胱尿液表现出显著富集的KEGG通路包括近端小管碳酸氢盐回收、离子通道、亚油酸代谢和肾素-血管紧张素系统。
关于尿路细菌微生态是如何促进泌尿系统结石的形成尚未有定论。一种可能的假设是细菌通过柠檬酸裂解酶的产生改变尿液的饱和度,从而降低尿液柠檬酸水平并导致晶体形成,随后细菌可能诱发炎症反应和促炎蛋白的释放,这些促炎蛋白形成结石基质内芯,并由晶体发展为结石。
尿失禁(UI)是较为常见的下尿路症状(LUTS),可分为急迫性尿失禁(UUI)、压力性尿失禁(SUI)、真性尿失禁及混合性尿失禁(mixed urinary incontinence,MUI)。现有研究表明,运用16s rRNA测序技术在患有UUI的成年女性中观察到了尿路细菌微生态的改变。UUI患者相较于非UUI患者的尿路菌群有更高丰度的加德纳菌( Gardnerella )和更低丰度的乳酸菌( Lactobacillus ),以及微生物多样性较低的UUI患者,其症状更为严重。一些乳酸菌(如卷曲乳酸菌)可能是UUI患者膀胱健康的标志,由于其具有产酸特性,可以通过控制无法在酸性环境中生存的细菌的生长来保护下尿路。亦有研究证实,尿路细菌微生态与UUI的发作、症状严重程度和治疗后罹患泌尿道感染(UTI)的风险相关。总之,迄今为止发表的研究已经证明尿路细菌微生态在UUI和对UUI治疗的反应中有明确的作用。未来对UUI患者的研究应致力于确定不同性别和年龄组之间的尿路细菌微生态差异是否与UUI的易感性有关。
膀胱过度活动症(over active bladder,OAB)是一种以尿急、伴或不伴急迫性尿失禁(UUI)为特征,通常伴有尿频和夜尿,并排除尿路感染(UTI)及其他相关病变的下尿路症候群。引发OAB的原因十分复杂,一些OAB症状是由神经肌肉和毒蕈碱受体的问题引发,但一些OAB患者对抗毒蕈碱受体、肉毒杆菌毒素或其他治疗没有反应。一项针对女性OAB患者的研究表明,非OAB人群的细菌多样性程度高于OAB人群,且OAB人群的微生物组表现出较大的差异性。在患病人群中检出最多的门是厚壁菌门,其次是变形菌门、放线菌门和拟杆菌门(图1-2-3-1)。在科水平上,OAB人群的双歧杆菌科( Bifidobacteriaceae )显著多于非OAB组。利用LEfSe算法识别与OAB相关的特定细菌属发现(图1-2-3-2),通过将对照组和OAB组划分为不同的分类,发现OAB类群中有13个属的丰度下降,包括普雷沃菌属( Prevotella )、小杆菌属( Dialister )、具核梭杆菌属( Fusobacterium )、乔奎泰拉菌属( Jonquetella )、弯曲菌杆属( Campylobacter )、大芬戈尔德菌属( Finegoldia )、厌氧球菌属( Anaerococcus )、乳酸杆菌属( lactobacillus )、锥体杆菌属( Pyramidobacter )、脲原体属( Ureaplasma )、肠球菌属( Enterococcus )、新鞘脂菌属( sphingobium )和乳球菌属( Lactococcus )。相比之下,OAB组中有7个属的相对丰度升高,包括纤毛菌属( Sneathia )、葡萄球菌属( Staphylococcus )、变形杆菌属( Proteus )、创伤球菌属( Helcococcus )、孪生球菌属( Gemella )、支原体属( Mycoplasma )和空气球菌属( Aerococcus )。该研究还发现,多样性较低的异常尿液菌群与较高水平的抑郁和焦虑呈正相关。这表明尿路菌群可能与肠道菌群一样具有与大脑沟通的潜力,特别是可能具有引发中枢敏化的潜能。因此,就像成熟的脑-肠道-微生态轴一样,脑-膀胱-微生态轴也可能存在。
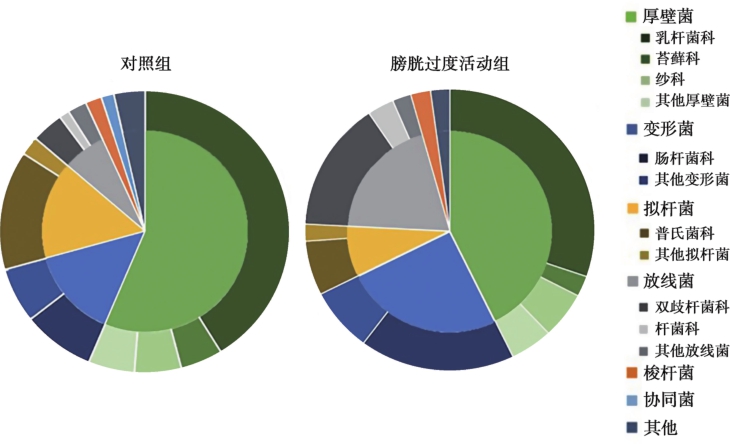
图1-2-3-1 比较对照组(左)和OAB患者(右)在门水平(内圈)和科水平(外圈)的分类差别
相对丰度<0.5%的细菌属被归类为“其他”。
总之,结合现有研究显示,OAB患者的尿路菌群较健康人群,在多样性及丰度上都显著下降,异常的尿路微生物群落可能在OAB的发病机制和治疗中有很强的指示作用。
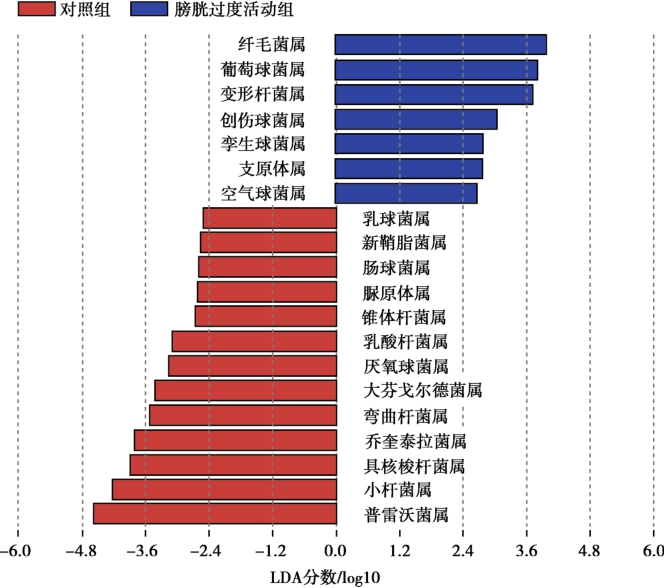
图1-2-3-2 OAB患者与对照组尿微生物组的LEfSe分析
红色为对照组富集菌属,蓝色为OAB患者富集菌属。只有符合线性判别分析评分阈值>2.5的属被展示。
利用高通量测序分析来自患间质性膀胱炎(IC)女性的清洁中段尿,结果显示,与无症状健康个体相比,在分类组成、丰度和多样性方面存在明显差异。尿液中乳酸杆菌属的丰度显著增加,整体丰度和生态多样性下降。乳酸杆菌通常与阴道微生物菌群有关,并可维持一个酸性环境,发挥保护作用,防止感染。新近的一项研究使用Ibis T-5000通用生物传感器系统技术分析了228名IC患者初始和中段尿液样本中的微生物(细菌和真菌),在尿液样本中发现了80多种不同的微生物物种(超过30个属)。虽然复发和非复发病例的菌种组成没有显著差异,但有症状复发的患者念珠菌和酵母菌等真菌的含量较高。
通过16s rRNA测序分析,与神经源性膀胱患者相比,膀胱功能正常的男性和女性患者也观察到了尿路细菌微生态的变化,健康人群的膀胱尿液样本显著富集乳酸杆菌属和棒状杆菌属,而其他细菌属,如克雷伯菌、肠球菌和大肠埃希菌主要存在于神经源性膀胱患者的尿液中。另一项对中段尿进行16s rRNA测序的研究表明,与对照组相比,CP/CPPS患者的尿路细菌微生态显示出更高的细菌多样性和梭状芽孢杆菌属的富集。综合相关研究表明,这些变化可能与下尿路症状严重程度、临床表型以及功能代谢途径紊乱有关。
(吴芃 编 郑波 审)
[1]THOMAS-WHITE K,BRADY M,WOLFE AJ,et al. The bladder is not sterile:History and current discoveries on the urinary microbiome[J]. Curr Bladder Dysfunct Rep,2016,11(1):18-24.
[2]BRUBAKER L,PUTONTI C,DONG Q,et al. The human urobiome[J]. Mamm Genome,2021,32(4):232-238.
[3]ARAGÓN IM,HERRERA-IMBRODA B,QUEIPO-ORTUÑO MI,et al. The urinary tract microbiome in health and disease[J]. Eur Urol Focus,2018,4(1):128-138.
[4]CAVARRETTA I,FERRARESE R,CAZZANIGA W,et al. The microbiome of the prostate tumor microenvironment[J].European Urology,2017,72(4):625-631.
[5]FENG Y,RAMNARINE VR,Bell R,et al. Metagenomic and metatranscriptomic analysis of human prostate microbiota from patients with prostate cancer[J]. BMC Genomics,2019,20(1):146.
[6]MIYAKE M,OHNISHI K,HORI S,et al. Infection and chronic inflammation in human prostate cancer:detection using prostatectomy and needle biopsy specimens[J]. Cells,2019,8(3):212.
[7]WANG J,LI X,WU X,et al. Uncovering the microbiota in renal cell carcinoma tissue using 16S rRNA gene sequencing[J].J Cancer Res Clin Oncol,2021,147(2):481-491.
[8]TANG R,JIANG Y,TAN A,et al. 16S rRNA gene sequencing reveals altered composition of gut microbiota in individuals with kidney stones[J]. Urolithiasis,2018,46(6):503-514.
[9]TICINESI A,MILANI C,GUERRA A,et al. Understanding the gut-kidney axis in nephrolithiasis:an analysis of the gut microbiota composition and functionality of stone formers[J]. Gut,2018,67(12):2097-2106.
[10]ZAMPINI A,NGUYEN AH,ROSE E,et al. Defining dysbiosis in patients with urolithiasis[J]. Sci Rep,2019,9(1):5425.
[11]DORNBIER RA,BAJIC P,VAN KUIKEN M,et al. The microbiome of calcium-based urinary stones[J]. Urolithiasis,2020,48(3):191-199.
[12]XIE J,HUANG JS,HUANG XJ,et al. Profiling the urinary microbiome in men with calcium-based kidney stones[J]. BMC Microbiology,2020,20(1):41.
[13]STERN JM,MOAZAMI S,QIU Y,et al. Evidence for a distinct gut microbiome in kidney stone formers compared to nonstone formers[J]. Urolithiasis,2016,44(5):399-407.
[14]KARSTENS L,ASQUITH M,DAVIN S,et al. Does the urinary microbiome play a role in urgency urinary incontinence and its severity?[J]. Front Cell Infect Microbiol,2016,6:78.
[15]WU P,CHEN Y,ZHAO J,et al. Urinary microbiome and psychological factors in women with overactive bladder[J].Frontiers in Cellular and Infection Microbiology,2017,7:488.