
下载掌阅APP,畅读海量书库
立即打开



我国结直肠癌(colorectal cancer,CRC)的发病率和死亡率保持上升趋势。2022年中国癌症统计报告显示:我国CRC发病率和死亡率分别位列第二位和第四位。与之相反,美国自2000年后在CRC发病率和死亡率方面呈现出逐年下降的趋势,这种差异主要与其癌症控制政策、健康生活方式建议、早癌筛查应用、治疗药物研发以及治疗策略改善相关。更加值得注意的是,我国CRC患者早诊率低,80%以上的患者在确诊时已属于中晚期,40%以上的患者发现时已发生肝肺转移。因此,CRC已成为严重威胁国民健康的重大疾病。
手术联合化疗或放疗是提高CRC患者生存的有效方法。近年来,随着EGFR抑制剂(如西妥昔单抗、帕尼单抗)、血管生成抑制剂(如贝伐珠单抗、雷莫芦单抗)和多激酶抑制剂(如瑞戈非尼、呋喹替尼)等靶向药物的发展,晚期CRC患者的预后得到进一步改善。此外,靶向 HER - 2 扩增、 BRAF V600E突变的药物也在临床研究阶段显示出积极的疗效。
在此基础上,以靶向PD-1/PD-L1通路、CTLA-4/B7-1通路为代表的免疫治疗进一步改变了CRC的治疗前景。继2015年首次报道微卫星高度不稳定(microsatellite instability-high,MSI-H)或错配修复缺陷(mismatch repair deficient,dMMR)是免疫检查点抑制剂(immune checkpoint inhibitor,ICI)治疗的敏感人群以来,后续多项研究也证实,ICI作为后线、一线及新辅助治疗在MSI-H/dMMR人群中均取得了突破性疗效。同时,基于ICI的双免联合、化疗免疫联合、靶向免疫联合及放疗免疫联合等方案在微卫星稳定(microsatellite stability,MSS)患者中也显示出了一定的疗效。随着人们对免疫微环境的深入研究,其他免疫治疗类型,包括过继细胞治疗(adoptive cell transfer therapy,ACT)、肿瘤疫苗、溶瘤病毒等,也在CRC临床研究中取得了突破疗效,值得进一步开发探索。
以上治疗手段的发展,为晚期CRC患者提供了更多的治疗选择。然而,如何将丰富的治疗手段转化为患者生存期的最大化延长,需要建立分子生物学特征指导下的CRC精准治疗体系。一方面,积极探索不同治疗手段(如免疫治疗与靶向治疗)的最佳联合模式,使更多的患者能够从联合治疗中获益;另一方面,开发出更加精准的生物标志物,实现治疗效果的预测和动态监测。当然,这与当前大力推广的以患者为中心的多学科诊疗模式是不谋而合的。
靶向药物是指被赋予了靶向能力的药物及制剂。相对于传统的化疗药物,靶向药物具有选择性好、毒副作用小的优点。传统的化疗药物利用细胞毒性抑制或杀伤快速分裂的细胞,如CRC中常用的奥沙利铂等铂类化疗药物、伊立替康等植物类化疗药物及氟尿嘧啶等抗代谢类化疗药物。但传统的化疗药物会同样损伤人体中分裂活跃的正常细胞,如造血细胞、黏膜上皮细胞等,进而产生骨髓抑制、胃肠道反应、肝肾损害等毒副作用。
随着肿瘤分子生物学的发展,越来越多的癌基因、肿瘤特异性抗原(tumor-specific antigen,TSA)和信号通路异常改变被发现。靶向药物通过靶向肿瘤相关的特异性分子靶点精准杀伤肿瘤细胞。根据分子性质,靶向药物可分为大分子类靶向药物和小分子类靶向药物。
大分子类靶向药物利用抗原抗体的特异性结合发挥作用,并根据分子结构可进一步分为单克隆抗体、双特异性抗体和单链抗体等。单克隆抗体是针对肿瘤细胞中一个特定的抗原表位产生的高度均一的抗体,如靶向表面生长因子受体的西妥昔单抗和靶向血管内皮生长因子的贝伐珠单抗。双特异性抗体在单克隆抗体的基础上增加了一个特异性抗原结合位点,可特异性结合两种不同的抗原或者同一抗原两个不同的抗原表位。作为一种工程化抗体,双特异性抗体具有单克隆抗体不具备的优势,如通过靶向两种肿瘤相关抗原(tumorassociated antigen,TAA)可以增强抗体的特异性、降低脱靶毒性;通过靶向两种肿瘤异常活化通路可以增强抗体效用;同时靶向TAA和免疫细胞相关抗原,可以增强免疫细胞对肿瘤细胞的杀伤能力等。单链抗体是由抗体重链可变区和轻链可变区通过15~20个氨基端的短肽连接而成的一种小分子基因工程抗体。由于去除了原有结构的Fc段并保留了抗原结合部位,具有分子量小、穿透能力强和免疫原性弱等优势。此外,单链抗体在嵌合抗原受体T细胞(chimeric antigen receptor T cell,CAR-T)治疗中亦得以应用。CAR-T的嵌合抗原受体由胞外区、跨膜区和胞内区3部分组成,其中单链抗体负责识别和结合胞外区目的抗原。
小分子类靶向药物通过竞争性抑制阻断激酶活性发挥作用,如酪氨酸酶抑制剂通过竞争性结合酪氨酸激酶阻断多种底物蛋白酪氨酸残基磷酸化进而抑制肿瘤增殖。
根据药物的分子性质,CRC靶向药物主要包括单克隆抗体和小分子抑制剂两大类。根据作用靶点的不同,目前被批准使用的CRC靶向药物主要可以分为靶向EGFR的单克隆抗体、靶向VEGF/VEGFR的单克隆抗体、抗血管生成的多靶点小分子抑制剂、靶向HER-2的单克隆抗体、靶向BRAF V600E的小分子抑制剂等。
EGFR属于表皮生长因子受体家族。该家族由ErbB1(EGFR/HER-1)、ErbB2(Neu/HER-2)、ErbB3(HER-3)和ErbB4(HER-4)4个成员组成。EGFR与EGF和TGF-α等配体结合后激活,由单体转化为同源或异源二聚体,进而激活RAS/RAF/MEK/ERK,PI3K-AKT和JAKSTAT3等下游信号通路,最终促使肿瘤的发生和进展。25%~77%的CRC存在EGFR过表达,并与预后不良显著相关。左侧CRC的EGFR表达高于右侧。
CRC中靶向EGFR的西妥昔单抗和帕尼单抗是FDA批准的转移性结直肠癌(metastatic colorectal cancer,mCRC)的一线用药,并被NCCN和ESMO指南建议在 BRAF 和 RAS 野生型的晚期CRC患者使用。西妥昔单抗和帕尼单抗与EGF和TGF-α等配体竞争性结合EFGR,阻断EGFR通路的激活,进而抑制肿瘤细胞增殖和转移、抑制肿瘤血管生成达到抑制肿瘤进展的目的。相对于鼠-人嵌合抗体的西妥昔单抗,完全人源化的帕尼单抗则不会引起抗体依赖的细胞介导的细胞毒性作用,超敏反应的发生风险也相对较低。但根据Ⅲ期ASPECCT研究显示,在难治性 KRAS 野生型的晚期CRC患者中,帕尼单抗和西妥昔单抗治疗后的总生存期(overall survival,OS)分别为10.4个月和10.0个月,差异无统计学意义。
血管生成受促血管生成因子和抗血管生成因子的复杂调控。肿瘤血管生成过程中,促血管生成因子异常过表达使得肿瘤血管异常生成、排列紊乱,进而促进肿瘤的发展和微环境的重塑。CRC中的抗血管生成的靶向药物包括靶向血管内皮生长因子/血管内皮生长因子受体的单克隆抗体和多靶点小分子酪氨酸激酶抑制剂。VEGF家族成员主要包括VEGF-A、VEGF-B、VEGF-C、VEGF-D、VEGF-E和胎盘生长因子(PIGF)等,其中VEGF-A在肿瘤血管生成中发挥重要作用。VEGFR包括VEGFR 1、VEGFR 2、VEGFR 3。VEGFR 1和VEGFR 2分布于肿瘤血管内皮细胞,调节肿瘤血管生成,其中VEGFR 2发挥主要作用;VEGFR 3则主要调节肿瘤淋巴管的生成。VEGFR 1在上皮细胞、炎性细胞和癌细胞中表达。具体功能尚不明确。目前认为VEGFR 1在上皮细胞的分化迁移、炎性细胞的激活和浸润中发挥关键作用。此外,VEGFR 1对VEGF-A的亲和力高于VEGFR 2,VEGFR 1与PIGF结合作用可以控制与VEGFR 2结合的VEGF-A的数量,从而参与VEGFR 2的血管生成调控。VEGFR 2主要与VEGF-A结合发挥作用,结合后胞内段的酪氨酸残基磷酸化,从而激活PLCγ、RAS/RAF/ERK/MAPK和PI3K-AKT等下游信号通路,最终导致肿瘤新生血管的生成和侵袭、转移的发生。在CRC中VEGF水平在早期阶段和晚期阶段均升高,在转移发生阶段升高尤为显著。其活性可由 KRAS 和 P53 突变、缺氧诱导因子等因素调控。
贝伐珠单抗是首个被FDA批准用于晚期CRC患者的第一个抗血管生成药物,靶向循环中VEGF-A发挥抗血管生成作用,在一线治疗、二线治疗和维持治疗中的有效性均得以证实。在一线治疗中,有效性不受 KRAS 基因状态的影响。左半和右半CRC对贝伐珠单抗的反应均良好。雷莫芦单抗是靶向VEGFR 2 的单克隆抗体,被批准使用于转移性CRC的二线治疗。阿柏西普可以抑制VEGF家族的多个成员,如VEGF-A、VEGF-B和PIGF,被批准用于转移性CRC的二线治疗。除单克隆抗体外,小分子酪氨酸酶抑制剂同样是抗肿瘤血管生成的有效手段。瑞戈非尼可抑制多种酪氨酸激酶的活性,如VEGFR、PDGFR、FGFR和BRAF等,被FDA批准用于标准化疗耐药的CRC。在一项Ⅲ期的临床研究中发现,与安慰剂相比,瑞戈非尼在难治性转移性CRC能显著延长患者的中位OS和无进展生存期(progression free survival,PFS)。呋喹替尼是由我国研发的靶向VEGFR 1、VEGFR 2和VEGFR 3的TKI。Ⅲ期FRESCO试验显示与安慰剂相比,呋喹替尼的OS(9.3个月vs 6.6个月, HR =0.65, P <0.001)和PFS(3.7个月vs 1.8个月, HR =0.26, P <0.001)显著延长。基于该结果,2018年,国家药品监督管理局批准呋喹替尼用于晚期难治性CRC。
位于染色体7q34的 BRAF 基因编码的丝氨酸/苏氨酸激酶是RAS/RAF/MEK/MAPK途径的关键调控分子。 BRAF 突变后使BRAF蛋白持续高度激活,使其不受上游RAS蛋白的调控,并不断激活下游MEK/MAPK通路,促进肿瘤细胞在增殖、代谢等方面异常改变。约60%的黑色素瘤存在 BRAF 突变,但CRC中 BRAF 突变比例不到10%,其中 BRAF V600E突变比例超过90%,且在女性和右半CRC中更加常见。 BRAF V600E突变是由于 BRAF 基因第15外显子的第1 799位核苷酸上胸腺嘧啶突变为腺嘌呤,其编码的氨基酸由缬氨酸变为谷氨酸,使 BRAF 活性调高约500倍。 BRAF V600E突变型和野生型mCRC标准化治疗后的中位OS分别为12~14个月和29~30个月。
靶向 BRAF 突变的多种小分子抑制剂已被FDA批准用于黑色素瘤的治疗,如维莫拉非尼、达拉非尼和恩哥拉非尼。但在难治性 BRAF 突变的CRC中,靶向 BRAF 突变的小分子抑制剂的反应率却仅为5%。 BRAF 突变性结直肠癌对维莫拉非尼的RAF抑制不敏感是由于EGFR介导的MAPK信号转导的再激活。多靶点治疗可能是 BRAF V600E的最佳解决方案。多项临床试验证实西妥昔单抗联合BRAF抑制剂的治疗效果优于BRAF单药治疗。靶向EGFR、BRAF和MEK的3药联合方案的反应率(21%)高于靶向EGFR、BRAF(10%)和靶向EGFR、MEK(0)的双药联合方案。BEACON试验提示,靶向EGFR、BRAF和MEK的3药联合方案的中位OS高于靶向EGFR、BRAF的双药治疗方案(9.0个月vs 8.0个月)。
HER - 2 与上述的EGFR同属于EGFR家族成员,故作用机制与EGFR相似。 HER - 2 与配体(目前尚不清楚)结合后,由单体转化为异源二聚体,进而激活RAS/RAF/MEK/ERK、PI3K-AKT和JAK-STAT3等下游信号通路,最终促使肿瘤的发生和进展。 HER - 2 扩增/过表达在乳腺癌(20%~30%)和胃癌(10%~15%)中常见,在晚期CRC中仅占2%~3%,并且与 RAS 或 RAF 突变无关。 HER - 2 扩增/过表达在左半CRC中更为常见。
目前CRC中,以 HER - 2 为靶点的靶向治疗适用于 KRAS 野生型 HER - 2 阳性的晚期CRC患者的二线治疗。抗EGFR治疗后全基因组血浆DNA测序提示, HER - 2 扩增/过表达可能与EGFR单抗耐药相关。临床前研究表明,EGFR阻断后, HER - 2 代偿性过表达,且 HER - 2 和EGFR的联合靶向治疗效果优于单一药物治疗。Ⅱ期HERACLES试验结果提示,抗HER-2治疗可能为抗EGFR耐药性结直肠癌提供新的选择:在难治性 KRAS 野生型 HER - 2 阳性的mCRC中,双重 HER - 2 阻断(曲妥珠单抗+拉帕替尼)后,总体缓解率达30%(8/27)。小样本病例报道提示在对曲妥珠单抗联合帕妥珠单抗获得性耐药的CRC患者中,曲妥珠单抗联合拉帕替尼的双重HER-2阻断方案可能同样适用。
RAS 癌基因家族包括3个人类基因 KRAS 、 NRAS 和 HRAS 。其基因编码产物为具有GTP水解酶活性的小型GTP结合蛋白。RAS蛋白可催化一系列丝氨酸/苏氨酸蛋白激酶的磷酸化并激活磷酸化的级联反应,激活MAPK、PI3K-AKT等信号通路,最终促进细胞的增殖、分化等。 RAS 基因突变后RAS蛋白持续处于活性状态,进而中断上游EGFR信号调节并持续激活下游信号。
RAS 基因突变约占CRC的一半,且与患者的不良预后显著相关。 KRAS 突变是 RAS 基因突变常见类型,约占CRC的45%,与EGFR靶向治疗耐药和不良预后相关。85%~90%的 KRAS 突变集中在2号外显子的第12、13密码子上;约5%发生在3号外显子的第62密码子上;约5%发生在4号外显子的第146密码子上。
尽管 KRAS 突变在CRC中较为常见且在CRC的诊断、治疗及预后中发挥关键作用,但是由于KRAS蛋白对GTP具有高亲和力以及缺乏理想的分子结合位点,直接靶向抑制KRAS的药物开发一直未取得成功。但近年来靶向 KRAS G12C 突变的两种小分子抑制剂AMG 510和MRTX849在 KRAS 突变的CRC临床试验中显示出较好的有效性和安全性。在一项Ⅰ期临床研究(NCT 03600883)中,AMG 510被用于多种 KRAS G12C 突变的实体瘤中。其中纳入的42例经标准治疗后进展的 KRAS G12C 突变CRC患者经AMG 510治疗后3例达到客观缓解、32例达到疾病控制,中位PFS为4个月。在一项Ⅰ/Ⅱ期多中心临床研究中,4例 KRAS G12C 突变的CRC患者接受MRTX849后,1例部分缓解并且耐受性好。
ALK蛋白由位于2号染色体的 ALK 基因编码,是一种受体酪氨酸激酶,属于胰岛素受体超家族。肝素和ALK结合后,激活下游的JAK-STAT、PI3K-AKT、MAPK等信号通路,调控细胞的生长、转化和凋亡。 ALK 基因重排是 ALK 基因最常见的变异形式,占CRC的0.05%~2.5%。CRC中,ALK融合的常见伴侣基因包括 EML4 、 SPTBN1 、 CAD 、 SMEK2 、 STRN 、 SENPF 、 MAPRE3 、 PRKAR1A 、 C2orf44 和 PPP1R21 等。融合基因编码的ALK融合蛋白无须与配体肝素结合即可持续激活下游通路,导致细胞的恶性转化、增殖和转移。
ALK 基因重排的发生率极低,大规模的临床试验较难展开,但是陆续有个案报道提示发生 ALK 基因重排的CRC患者可从ALK靶向治疗中获益,如 STRN - ALK 基因重排和 CAD - ALK 基因重排等。此外,除 ALK 基因重排外,CRC中约有3.4%的患者发生 ALK 基因拷贝数增加或扩增。 ALK 扩增与患者不良预后相关,且对EGFR单抗治疗不应答。针对 ALK 基因扩增的CRC患者能否从靶向治疗中获益尚无定论,亟待进一步研究。
CRC中, ROS1 融合报道较少,仅有1例 SLC34A2 - ROS1 融合和 GOPC - ROS1 融合的个案报道。由于 ALK 和 ROS1 的激酶活性区域有70%的相似性,ALK抑制剂有望用于 ROS1 基因重排mCRC患者的治疗。
神经营养因子受体酪氨酸激酶(neurotrophin receptor kinase,NTRK)基因家族有 NTRK1 、 NTRK2 、 NTRK3 三个成员,分别位于染色体1q22、9q21、15q25不同区段,并分别编码TRKA、TRKB和TRKC三种原肌球蛋白受体激酶家族蛋白。TRK与相应的配体结合后,自身发生二聚化和磷酸化,并激活MAPK、PLCγ、PI3K-AKT等下游信号通路,调控细胞的增殖、分化、代谢、凋亡。 NTRK 基因重排后,TRK融合蛋白处于持续激活状态,引发下游信号通路级联反应,驱动肿瘤发生。
在CRC中 NTRK 基因重排的比例为0.2%~2.4%,常见融合有 TPM3 - NTRK1 和 LMNA - NTRK1 。2017年美国临床肿瘤学会(American Society of Clinical Oncology,ASCO)和2018年欧洲肿瘤内科学会(European Society for Medical Oncology,ESMO)年会报道了NTRK靶向药物拉罗替尼针对 NTRK 基因融合的肿瘤患者具有较好的疗效。恩曲替尼和拉罗替尼被FDA批准用于 NTRK 融合的mCRC。
ALK 、 ROS 和 NTRK 基因重排的CRC患者预后较差,对EGFR单抗治疗原发性不应答,部分解释了EGFR单抗在右半结肠 RAS/BRAF 野生型患者中获益有限的原因。因此,对存在 ALK 、 ROS 或 NTRK 基因重排的右半结肠病变的患者,除了考虑对应的靶向治疗,一线治疗中使用强化治疗方案如FOLFOXIRI联合贝伐珠单抗,可能也是比较合理的选择。
RET 基因融合发生在约0.4%的CRC患者,在右半CRC、 RAS/RADF 野生型和MSI-H的CRC患者中更加常见,且与患者的不良预后相关。存在 RET 基因融合的mCRC患者预后更差,平均OS约14个月。目前已有研究显示,多靶点抑制剂普纳替尼和凡德他尼在肠癌 RET 基因融合的人源肿瘤异种移植模型中取得良好效果。
由 MET 原癌基因编码的肝细胞生长因子(HGF)的受体酪氨酸激酶在肿瘤增殖、存活、转移和获得性耐药性中起着至关重要的作用。 MET 突变和扩增在CRC患者中很少发现,其发生率分别为2%~5%和0.5%~2.0%。尽管已有靶向 MET 的多种抑制剂,但是还未获批用于CRC。
FGFR2 扩增发生在约5%的胃癌中,目前应用NGS技术也发现CRC患者中存在 FGFR 扩增,但具体比例不详,FGFR/STAT通路是这类患者的治疗靶点。 EGFR 突变在肠癌中很少发生,多发生于EGFR单抗西妥昔继发性耐药的患者中。经西妥昔单抗治疗后,一部分患者EGFR胞外段发生突变(Ser492),阻止了西妥昔单抗和EGFR胞外段的有效结合,下游激活信号通路不能被西妥昔单抗抑制,导致肿瘤发生进展。而 EGFR Ser492 突变不影响帕尼单抗和EGFR结合并对下游EGFR通路发挥抑制作用,因此,此类突变患者换用帕尼单抗仍然能获得较好的疗效。 CDK 基因扩增是癌症中最常见的改变之一,在肿瘤中的发生率为15%~40%,有研究表明,在CRC患者中也存在 CDK 扩增,但具体比例尚不明确。CDK抑制因子通过靶向细胞周期蛋白,进而阻止肿瘤生长。尽管目前在CRC中开展的研究报道较少,但CDK4/6抑制因子在乳腺癌中成功应用将会对包括CRC在内的许多其他实体肿瘤的治疗产生深远影响。
(卢瑗瑗 聂勇战)
机体肿瘤免疫的过程包括肿瘤抗原释放和提呈、效应性T细胞启动和激活、T细胞向肿瘤组织迁移浸润、T细胞识别杀伤肿瘤细胞等4个过程。肿瘤免疫的正常进行是肿瘤抗原识别后,共刺激因子和肿瘤免疫相关抑制因素相互作用和平衡的结果。
免疫检查点是指免疫系统中存在的一些抑制性信号通路,机体在正常抗肿瘤免疫应答情况下,共刺激信号和共抑制信号保持平衡,通过调节自身免疫反应的强度来维持免疫耐受。机体在受到肿瘤侵袭时,通常会阻断免疫检查点信号通路从而抑制自身免疫,给肿瘤细胞的生长和逃逸提供机会。CRC中常见的免疫检查点抑制剂包括PD-1抑制剂和CLTA-4抑制剂,分别靶向PD-1/PD-L1和CTLA-4/B7-1两个免疫检查点通路。
PD-1又称为CD279,属于免疫球蛋白基因超家族的成员,是表达于免疫细胞细胞膜的含有288个氨基酸的跨膜糖蛋白。PD-1的配体PD-L1又称为CD274,是B7家族成员之一,表达在肿瘤细胞和浸润边缘的部分或某些免疫细胞中。T细胞受体的长时间激活、T细胞分泌的促炎性细胞因子(如IFN-γ和TNF-α)可以上调肿瘤细胞表面PD-L1的表达。PD-1和PD-L1的结合促使PD-L1胞内段ITIM一级Src家族激酶ITSM的磷酸化,进一步激活SHP-2、SHP-1从而抑制T细胞的增殖、激活和IFN-γ的释放以防T细胞持续异常激活。靶向PD-1/PD-L1的ICI主要在T细胞识别杀伤肿瘤细胞的过程中重新激活T细胞并增加其杀伤能力。
CTLA-4又称CD152,是由 CTLA - 4 基因编码的一种跨膜蛋白质,表达于活化的CD8 + 和CD4 + T细胞。CTLA-4与T细胞表面的协同刺激分子受体(CD28)具有高度同源性,并与相同的配体CD86(B7-2)和CD80(B7-1)结合。细胞表面的CTLA-4竞争性抑制CD28与抗原提呈细胞表面的B7蛋白(CD80、CD86)结合介导的共刺激信号,从而抑制CD8 + T细胞的免疫活性;另一方面,在Treg细胞中,CTLA-4能提高Treg细胞对CD8 + T细胞的抑制作用。靶向CTLA-4的ICI主要在肿瘤抗原提呈、效应性T细胞启动和激活及免疫微环境调节中发挥作用。
基于KEYNOTE016和Checkmate-142的结果,ICI被批准用于dMMR/MSI-H mCRC的二线治疗。KEYNOTE016(NCT 01876511)是一项Ⅱ期临床试验,以探究使用帕博利珠单抗在难治性mCRC患者中的疗效及安全性。结果发现在dMMR/MSI-H mCRC患者中,客观缓解率(object response rate,ORR)和疾病控制率(disease control rate,DCR)分别为50%和89%;在错配修复正常(mismatch repair proficient,pMMR)和微卫星低度不稳定(microsatellite instability-low,MSI-L)的mCRC患者中,ORR和DCR分别为0和16%。Checkmate142(NCT 02060188)探究了纳武利尤单抗(联合或不联合伊匹木单抗)在dMMR/MSI-H mCRC患者中作为二线治疗的疗效及安全性。纳武利尤单抗单药治疗队列中,中位随访1年的PFS为50%、OS为73%,中位反应持续时间尚未到达。两药联合中位随访时间为13.4个月时,ORR、完全缓解(complete response,CR)率和部分缓解(partial response,PR)率分别为49%、4%和45%。
KEYNOTE177是一项国际性随机对照临床研究,聚焦于评估帕博利珠单抗作为dMMR/MSI-H mCRC一线治疗的疗效及安全性。基于KEYNOTE177的结果,帕博利珠单抗被批准使用于dMMR/MSI-H mCRC患者的一线治疗。帕博利珠单抗治疗组的PFS和ORR分别为16.5个月和43.8%。标准治疗组的PFS和ORR分别为8.2个月和33.1%,且帕博利珠治疗组的副作用更少。
ICI在dMMR/MSI-H mCRC患者中的治疗作用显著。进一步的临床探索主要围绕“dMMR/MSI-H CRC患者中治疗前移”及“pMMR/MSI-H mCRC患者中,利用联合治疗策略突破ICI原发耐药”两个方向展开。
Checkmate142研究除探究免疫检查点抑制剂作为dMMR/MSI-H mCRC患者二线治疗的疗效外,还探索了双ICI联合作为一线治疗在dMMR/MSI-H mCRC患者的疗效。中位随访时间为13.8个月时,ORR、DCR和 CR 分别为60%、84%和7%。当中位随访时间为29个月时,DCR仍为84%,但是 CR 从13.8个月时的7%增长到13%,ORR从60%增长到69%。值得一提的是,相比KEYNOTE177中采用的帕博利珠单抗的单药治疗,纳武利尤单抗和伊匹木单抗联用有更好的疗效和安全性。
COMMIT(NCT 02997228)是一项Ⅲ期临床研究。在347例dMMR/MSI-H mCRC患者中探索mFOLFOX6/贝伐珠单抗联合不联合阿特珠单抗的疗效。目前尚未有结果报告。
NICHE(NCT 03026140)是一项正在进行的Ⅱ期探索性研究。该研究共纳入40例Ⅰ~Ⅲ期的结肠癌患者,其中包括可接受切除的21例dMMR结肠癌患者和20例pMMR结肠癌患者。dMMR结肠癌患者术前接受1剂伊匹木单抗和2剂纳武利尤单抗治疗。治疗结果显示:20例dMMR结肠癌患者均到达PR,19例实现主要病理缓解(major pathologic response,MPR),12例实现病理学完全缓解(pathologic complete response,pCR);而pMMR组也达到27%(4/15)的病理缓解率和20%(3/15)的主要病理缓解率。该结果初步证实了ICI作为新辅助治疗的有效性。
VOLTAGE(NCT 02948348)是一项Ⅰb/Ⅱ期单臂前瞻性研究,以研究放化疗后序贯纳武利尤单抗在局部进展期直肠癌中作为新辅助治疗的疗效。VOLTAGE-A分为包含37例MSS患者的A1队列和包含5例MSI-H患者的A2队列。结果显示,队列A1中有11例实现了PCR,14例取得了MPR。队列A2中,3例到达pCR。
pMMR/MSI-L CRC约占所有CRC患者的95%,为了打破高达95%的CRC患者对ICI原发耐药的现状,研究者探索了ICI与多种治疗手段联合的策略,以打破其免疫抑制的状态,使更多的免疫细胞进入肿瘤微环境,增强ICI的疗效。
(1)双靶点ICI联合:
研究者证明针对不同靶点的ICI联合具有协同抗肿瘤的效果。CCTG CO.26(NCT 02870920)是一项Ⅱ期临床试验,旨在研究在pMMR/MSI-L CRC患者中,与最佳支持治疗相比,靶向PD-L1的度伐利尤单抗和靶向CTLA-4的替西木单抗联合应用的疗效和安全性。结果表明双药联合使中位OS延长了2.5个月(6.6个月vs 4.1个月)。这也是首项证明双药联合优于最佳支持治疗的临床试验。
(2)ICI联合放疗:
研究表明,放射治疗(radical therapy,RT)会导致免疫原性细胞死亡,引起损伤相关分子模式,从而增加抗原提呈,促进T细胞的激活和浸润。损伤相关分子模式释放的分子有癌症相关抗原、炎症因子等。早期临床试验中,ICI联合放疗的治疗效果不尽如人意。一项单臂Ⅱ期临床试验(NCT 02437071)表明,在22例接受免疫检查点抑制剂联合放疗的pMMR/MSI-L CRC患者中,仅有1例有效。近年来鼓舞人心的结果日益增多。一项Ⅱ期临床试验(NCT 03104439)中,24例pMMR/MSI-L CRC患者接受双免疫抑制剂联合放疗后,DCR和ORR分别是29.2%(7/24)和12.5%(3/24)。VOLTAGE-A1是一项Ⅰ/Ⅱ期临床研究,纳入的37例MSS局部进展期直肠癌患者在接受放化疗序贯ICI的新辅助化疗后,11例达到了pCR,3例为病理退缩分级1级,14例取得了MPR。
(3)ICI联合MEK抑制剂:
MEK是RAS-MAPK通路的下游效应分子,临床前研究数据表明,抑制MEK可以增加癌症相关性抗原和PD-L1的表达,继而促进T细胞的浸润,从而增加ICI的抗肿瘤活性。一项Ⅰb期的临床研究(NCT 01988896)首先尝试MEK抑制剂考比替尼和PD-L1抑制剂阿特珠单抗的联合治疗策略。该项研究在2018年最终结果提示84例患者中仅7例有部分反应。虽然该结果说明MEK抑制剂与ICI有一定的作用,但是随后第二阶段的Ⅲ期临床对照研究IMBlaze370无有效事件出现。
(4)ICI联合抗血管生成剂:
抗血管生成剂通过多种途径增加ICI的效用,如增加CD8 + T细胞的浸润;上调肿瘤细胞PD-L1的表达;减少免疫抑制细胞浸润,如TAMS和Treg细胞;增强抗原提呈细胞的提呈作用等。一项Ⅰb期临床研究中,纳入的14例pMMR/MSI-L rCRC患者接受贝伐珠单抗联用阿特珠单抗的策略,研究效果显示,14例患者中有1例患者实现OR,9例患者病情稳定。REGONIVO研究(NCT 03406871)得到了同样可喜的结果,该研究纳入25例CRC患者(其中24例为pMMR/MSS,1例为dMMR/MSI-H)用以检测纳武利尤单抗联合瑞戈非尼的安全性和有效性,结果显示,ORR为33.3%,中位PFS为7.9个月,12个月的PFS和OS分别为41.8%和68.0%。基于此可喜结果,研究者正在扩大队列以进一步明确疗效。
(5)ICI联合双特异性抗体:
双特异性抗体能够靶向肿瘤细胞和T细胞上的两种不同的抗原,桥接肿瘤细胞和T细胞,进而促进T细胞的激活并增加T细胞的浸润。CEA-CD3是首个被报道的对MSS CRC患者有效的双特异性抗体。NCT 02650713结果显示,11例MSS CRC患者经过阿特珠单抗联合CEA-CD3治疗后,2例达到PR,其余9例达到DCR。除CEA-CD3外,双特异性抗体TRAILR2-CDH17、鸟苷酸环化酶C(guanylyl cyclase C,GUCY2C/GCC)CD3和CD137-PD-L1在临床前研究中显示出了较好的抗肿瘤特性。
(6)ICI的其他联合治疗策略:
除上述治疗策略外,仍有一些联合治疗的策略值得关注,如联合肿瘤疫苗、溶瘤病毒和粪菌移植。其中溶瘤病毒通过介导肿瘤免疫原性细胞死亡诱发抗肿瘤免疫,从而使“冷肿瘤”转为“热肿瘤”,以增强ICI的抗肿瘤效应。另外肠道菌群作用显著,如假长双歧杆菌可以通过释放肌酐促进T细胞活性,进而增强ICI的抗肿瘤疗效。
错配修复(mismatch repair,MMR)是维持DNA保真性的重要手段之一。根据错配修复蛋白缺失状态,CRC分为pMMR型和dMMR型两种。MMR异常引起的插入和缺失突变可以引起微卫星长度的改变。根据微卫星位点的稳定状态,将CRC分为MSS、MSI-L和MSI-H三种。其中,MSS中的5个微卫星位点均为稳定状态,MSI-L中有且仅有1个卫星位点不稳定,MSI-H中存在2个及2个以上不稳定的微卫星位点。
不同的错配修复状态多与相应的微卫星不稳定状态相匹配。综合错配修复和微卫星状态,可以将CRC分为dMMR/MSI-H CRC和pMMR/MSS/MSI-L CRC两种。其中约15%的CRC患者为dMMR/MSI-H CRC,且dMMR/MSI-H CRC所占比例随着肿瘤分期的增加而减小。Ⅱ期、Ⅲ期和Ⅳ期的dMMR/MSI-H型分别占相应分期患者总数的5%~20%、11%和5%。dMMR/MSI-H能一定程度预测不同分期的CRC患者的预后。Ⅱ期和Ⅲ期的dMMR/MSI-H CRC患者预后好于pMMR/MSI-L CRC。此外,尽管Ⅳ期dMMR/MSI-H CRC患者的预后明显差于pMMR/MSI-L CRC,却对ICI有更好的临床反应。
在dMMR/MSI-H CRC中,主要组织相容性复合体(major histocompatibility complex,MHC)Ⅰ类肽包括可被肿瘤免疫细胞识别的新抗原,故肿瘤微环境中有更多的CD8 + 肿瘤浸润淋巴细胞、T辅助1(TH1)、CD4 + T细胞和巨噬细胞浸润。同时,dMMR/MSI-H肿瘤细胞的T细胞抑制配体明显上调以促进肿瘤细胞免疫逃逸,如B7家族的PD-L1、CD80和CD86等。相反,因为pMMR/MSI-L CRC中MHC Ⅰ类分子几乎与正常细胞无差异,故免疫细胞浸润少,T细胞抑制配体表达量少,继而导致ICI的治疗效果差。
肿瘤突变负荷(tumor mutational burden,TMB)是多种实体瘤疗效的独立预测因子。尽管MSI-H肿瘤的TMB值亦高,但是TMB值高的肿瘤中仍然包括部分MSS的肿瘤。REGONIVO的研究者评估了23例CRC患者的TMB状态并根据TMB高低水平分组后发现:TMB高水平组和低水平组的ORR分别为50%和35.3%,中位PFS分别为12.5个月和7.9个月。在CCTG CO.26中,研究者通过分析血清中的cfDNA评估TMB水平,将28作为“cut off”值将患者分为TMB高低两组。发现TMB高水平组有更好的OS。TMB是非常有希望的生物标志物,但是仍需更多的数据支持。
POLE和POLD1对于DNA复制中的校对和保真度至关重要。POLE和POLD1的生殖细胞或体细胞突变会使基因校准功能异常从而导致肿瘤的发生并且增强肿瘤细胞的免疫原性。据统计,约有7.4%的CRC患者携带 POLE 或 POLD1 的突变,MSS或MSI-L的肿瘤约占携带 POLE 或 POLD1 突变肿瘤的74%。在pMMR CRC中, POLE 突变的CRC具有更多的CD8 + T细胞浸润,并且免疫检查点的表达水平更高。利用POLE或POLD-1的表达水平差异有望进一步将MSS CRC中可能从ICI治疗获益的人群筛选出来。目前探究 POLE 突变在ICI治疗中的指导性作用的临床研究有NCT 03435107、NCT 03827044和NCT 03150706。
肿瘤浸润淋巴细胞(tumor infiltrating lymphocyte,TIL)与CRC患者预后相关。相对于TNM分期,TIL具有更好的预后指示作用。免疫评分根据肿瘤边缘及肿瘤中心的CD3 + T细胞和CD8 + T细胞的密度进一步将肿瘤分为冷肿瘤、热肿瘤。
尽管PD-L1是目前ICI的重要靶点,但PD-L1的水平尚未证明与ICI的治疗效果相关。KEYNOTE106的后续分析提示PD-L1的表达水平与ICI治疗后的PFS和OS无关。Checkmate142的后续分析也提示PD-L1的表达水平与治疗效果之间无相关性。
肠道菌群与多种实体瘤中的ICI疗效相关。研究证明,假长双歧杆菌、阿克曼菌、约氏乳杆菌等能够增强ICI的疗效,这说明肠道菌群的组成有望成为ICI治疗的预测标志物。
在晚期CRC中,ICI仅表现出对MSI-H/dMMR患者的临床应答,而这部分患者仅占比5%,这意味着大多数CRC患者无法从中获益。一部分研究认为ICI治疗失败是由于肿瘤局部缺乏充分的淋巴细胞浸润,ACT作为一种极具潜力的免疫治疗手段,或许能从机制上弥补ICI治疗的缺陷,从而扩大免疫治疗在CRC患者中的受益人群。ACT的发现可追溯至20世纪50年代,伴随着移植物抗肿瘤反应的发现以及T细胞在此过程中所起的关键作用,促使人们开始积极探索T细胞在抗肿瘤免疫中的作用。20世纪80年代Rosenberg团队首次从多种小鼠肿瘤模型中分离出TIL并证明了其在小鼠荷瘤模型中的治疗效果,为TIL治疗恶性肿瘤的临床应用提供了理论基础。然而,TIL本身存在的体外长期培养易衰竭、体内无法长期存活等特点,导致接受TIL治疗的临床试验的患者退出率超过50%,极大地阻碍了其临床应用。而在过去的20年中,随着基因编辑和高通量测序技术的出现和广泛应用,推动了更为切实可行的肿瘤反应性淋巴细胞生产方案,ACT又逐渐进入人们的视野并成为癌症免疫治疗中最有前景的策略之一。目前CAR-T、T细胞受体-工程化T细胞(T cell receptor-T cell,TCR-T)和TIL等疗法已逐渐应用于CRC的治疗中。
ACT是指通过从患者体内收集免疫活性细胞,在体外进行扩增、基因工程改造和功能鉴定,再回输到患者体内达到直接杀伤肿瘤细胞或激发机体的免疫应答杀伤肿瘤细胞的目的。根据是否具有明确的免疫细胞靶点,ACT可分为特异性免疫细胞治疗和非特异性免疫细胞治疗两大类。其中,特异性免疫细胞治疗包括在经历基因工程改造后提高了细胞对肿瘤抗原的识别能力,可以特异性杀伤肿瘤细胞。除了已广泛应用于临床的CAR-T细胞治疗,近年来基因工程改造的TCR-T、TIL、嵌合抗原受体NK细胞(CAR-NK)及嵌合抗原受体巨噬细胞(CAR-M)治疗等新型ACT也开始逐渐进入临床。而自体淋巴因子激活的杀伤细胞、自体树突状细胞-细胞因子诱导杀伤细胞(dendritic cell-cytokine induced killer cell,DC-CIK)等非特异性免疫细胞治疗,由于其在临床应用中未表现出良好的治疗疗效,已逐渐被淘汰。
嵌合抗原受体(chimeric antigen receptor,CAR)是一种模块化、基因改造合成的抗原受体,具有抗体特性及高效的TCR激活信号。CAR的结构中有4个主要组件,分别是抗原结合域、铰链区、跨膜结构域和胞内信号结构域。抗原结合域传统上是由可变重链(VH)、可变轻链(VL)及两者之间的柔性连接物形成的单链可变片段(single-chain variable fragment,scFv),抗原结合域可识别肿瘤抗原,从而实现独立于MHC的细胞活化;铰链区能提供足够的灵活性来克服空间位阻,以便结合靶抗原;跨膜结构域负责将CAR结构锚定在所转导细胞膜上;胞内结构域通常包括激活区(信号转导结构域)、一个或多个共刺激结构域,信号转导结构域可为细胞增殖活化提供第一信号,共刺激结构域则为细胞的增殖活化提供第二信号。
在CAR相关细胞治疗中,CAR-T是应用最多且技术相对成熟的一种治疗方法。CAR-T是指通过基因修饰技术,将带有特异性抗原识别结构域及T细胞激活信号的CAR结构转入T细胞,使T细胞能以非MHC限制的方式直接与肿瘤细胞表面特异性抗原结合而被激活,释放穿孔素、颗粒酶B等杀伤肿瘤细胞,同时可释放细胞因子募集人体内源性免疫细胞。CAR-T的主要优势在于以非MHC限制的方式与癌细胞结合,使更多的癌细胞易受到攻击,且与传统化疗药物等相比,CAR-T可有效杀伤细胞表面抗原表达量较低的肿瘤细胞,还可形成免疫记忆T细胞,获得长效的特异性抗肿瘤效应。但CAR-T通常仅识别肿瘤细胞表面表达的特异性抗原,因此能够作为CAR-T靶标的抗原相对局限,并且多数CAR-T细胞单个肿瘤抗原靶点的特性可致肿瘤产生抵抗发生抗原逃逸。除此之外,CAR-T在实体瘤中的应用也受到其脱靶效应、局限性肿瘤细胞外附着、免疫抑制微环境对CAR-T细胞的抑制效应,以及治疗毒性,如细胞因子释放综合征(cytokine release syndrome,CRS)、CAR-T相关性脑病综合征等的限制。因此,目前研究人员主要考虑的途径是实现逻辑门控CAR和可控制的CAR。逻辑门控CAR是设想实现CAR的特异性多蛋白识别,使CAR-T细胞能更精确靶向癌细胞并减少抗原逃逸,可供选择的策略基于逻辑门控和条件表达系统:针对特定肿瘤抗原CAR的表达依赖于另一种肿瘤抗原受体(如synNotch受体)的激活,必须结合多个抗原才能完成CAR-T细胞激活。对于可控制CAR的实现,目前可通过连接CD3ζ和共刺激域分离的受体,即一个受体具有CD3ζ激活域,第二个受体具有共刺激域,每个受体识别不同的抗原,只有识别到多抗原才能完成CAR-T细胞的活化。对于可控制CAR,部分设想为在特定刺激(如超声、蓝光)下CAR才可激活并表达于T细胞表面,在无刺激情况下,CAR-T细胞则不具备细胞毒性,但这一研究领域目前还尚在初期,许多研究处于概念验证阶段。
除CAR-T细胞治疗外,CAR转导技术还可应用于自然杀伤细胞(natural killer cell,NK)、巨噬细胞等免疫细胞类型中。其中CAR-NK是指将NK细胞经CAR基因修饰,赋予其靶向识别肿瘤细胞的能力。与CAR-T细胞相比,CAR-NK细胞的寿命短,在血液循环中停留时间短,无明显的移植物抗宿主反应,临床上显示出更好的适应性与安全性;此外,活跃的NK细胞主要产生IFN-γ和粒细胞-巨噬细胞集落刺激因子,对机体无严重毒性影响。在目前的研究中还发现实体瘤对CAR-NK细胞治疗更为敏感。
CAR-M治疗则是将经过基因编辑的特定CAR基因植入巨噬细胞内,在CAR结构识别结合肿瘤细胞表面特异性抗原后,CAR-M激活并吞噬肿瘤细胞。除吞噬杀伤作用外,CAR-M细胞还可释放细胞因子和趋化因子促进DC的抗原提呈能力。与CAR-T相比,CAR-M存在诸多优势,如实体瘤微环境渗透、激活适应性免疫应答,以及在恶性实体瘤环境中生存,因此CAR-M得以在实体瘤治疗领域中显示出较好的治疗效果。
在体内,T细胞表面的TCR可通过与HLA的结合来识别异常的蛋白片段,从而对靶细胞发起攻击并杀灭靶细胞。获取某些识别特定肿瘤抗原的TCR克隆并转染至自体T细胞,从而使这些转染细胞具备对肿瘤的特异性杀伤能力。TCR-T正是基于这一原理,通过筛选和鉴定能够特异性结合肿瘤抗原的TCR,将该段基因转染至肿瘤患者自体T细胞后回输至患者体内,从而达到治疗肿瘤的目的,目前克隆转导的TCR序列主要来自α βT细胞。第一代TCR-T是从患者T细胞中分离出肿瘤抗原特异性识别的T细胞亚群,经体外扩增后回输治疗。由于这种T细胞克隆数量极少,个体差异很大,因此很难产业化。第二代TCR-T是通过克隆上述肿瘤抗原特异性识别的T细胞,获取其TCR基因序列,再转导至患者的外周T细胞,这种方法使得TCR-T产业化成为可能。第三代TCR-T则是通过优化TCR的亲和力,使其能够更好地识别肿瘤抗原,再将其转导至患者T细胞,整体提高TCR-T的成药性。第四代TCR-T是靶向肿瘤新抗原(neoantigen)的高特异性细胞疗法,肿瘤应答和安全性大幅提高,但由于个体突变差异,治疗可及性还有待进一步研究。同时,目前还有通用型TCR-T以及基于γδ T细胞受体的新型癌症免疫疗法,γδ T细胞的TCR相比更常见的α βT细胞能更好地识别癌细胞的压力信号,进而使γδ T细胞TCR只允许杀死那些有癌症压力信号的细胞,在安全性上可得到进一步完善。
与CAR-T细胞只能识别肿瘤细胞表面表达的TSA不同,TCR-T识别MHC所提呈抗原,不受抗原是否表达在肿瘤细胞表面的限制,也可识别细胞内抗原或肿瘤新抗原,对肿瘤细胞的精准靶向性更强;此外,TCR-T更容易向实体瘤内部渗透,而CAR-T通常在肿瘤外部附着,不易向内部渗透;由于TCR-T特异性表达于T细胞表面TCR结构,本身来源于正常的人体,不易引起机体的免疫排斥,抗抗体产生的概率低,而CAR-T中CAR结构是人为改造的基因,机体对CAR的排斥会较强,可能会缩短CAR-T的存活时间。同时,TCR-T细胞同样具有免疫记忆功能,可在体内存活较长时间,带来长期的特异性抗肿瘤效应。但TCR-T细胞治疗技术在抗肿瘤治疗疗效的完善中仍面临几个关键挑战,如治疗性TCR亲和力的增加、共有TSA和TCR的鉴定、TCR表达的调节。这些难点的解决将有助于充分发挥TCR-T细胞治疗的潜力,在抗肿瘤治疗中发挥更大的作用。
TIL治疗是指从肿瘤组织中分离肿瘤浸润淋巴细胞,在体外培养和大量扩增后回输到患者体内的技术。其原理是通过确定患者体内肿瘤细胞的特定突变,利用突变信息找到能有效靶向这些突变进行打击的淋巴细胞,提取并经体外培养扩增等操作后,重新输入患者体内,发挥抗肿瘤作用。TIL治疗中所分离的淋巴细胞来源于肿瘤组织,识别肿瘤的能力高于血液来源的淋巴细胞,而且是经过天然选择与富集、肿瘤特异性T细胞比例高且多样性丰富的群体,具有多靶点、肿瘤趋向、浸润能力强、副作用小等优点。自TIL问世以来,最常用于黑色素瘤的治疗,除此之外,也有研究观察了TIL在宫颈癌、卵巢癌、肾癌、胃肠道肿瘤、头颈部鳞癌中的治疗疗效,但结果并不显著。
与CAR-T和TCR-T细胞治疗相比,TIL治疗存在一定的优势。首先,TIL是肿瘤组织中分离的肿瘤浸润淋巴细胞,因此可以通过多个靶点激发对肿瘤细胞的细胞毒性反应。其次,由于TIL治疗使用的细胞是经过天然选择与富集、肿瘤特异性T细胞比例高且多样性丰富的群体,尚未观察到其脱靶效应与CRS的出现,因此TIL较CAR-T和TCR-T安全性更高。另外,TIL的关键点在于体外扩增T细胞的数量与质量,无须经过基因编辑修饰与转导,因此成本相对较低。但该技术同样存在一定的缺陷,由于TIL体外培养条件要求高,培养时间长,需要新鲜的肿瘤组织、癌性胸腔积液或转移淋巴结,而且并非所有患者标本均可培养出TIL,因此,其临床应用有待进一步研究。
除此之外,ACT还包括自体淋巴因子激活的杀伤细胞、细胞因子诱导的杀伤细胞(cytokine-induced killer cell,CIK)等非特异性细胞治疗。但随着基因工程技术的发展,非特异性疗法由于细胞靶向的非特异性、治疗所带来的诸多不良反应以及临床实践中未表现出良好的疗效,已逐渐被淘汰(表9-2-1)。
表9-2-1 不同ACT细胞治疗间的对比
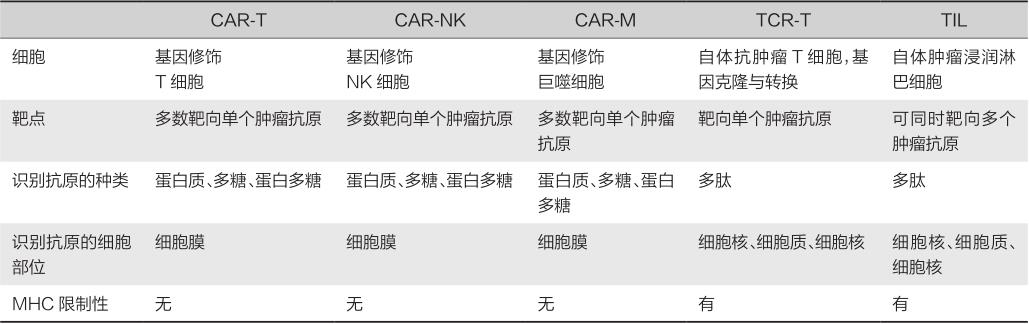
注:MHC.主要组织相容性复合体。
在过去20年中,ACT在B细胞白血病和淋巴瘤等类型的癌症中取得了显著成功。ACT在实体瘤的探索要追溯到1988年,Rosenberg团队首次在晚期黑色素瘤患者中观察到ACT具有一定的治疗效果。从那时起,人们在多种实体瘤中对ACT疗效展开了深入的研究,凭借主动免疫具有的显著优点,ACT目前正成为癌症免疫治疗中最有前景的策略之一。目前正在开发用于CRC的ACT主要有3种形式,包括TIL、CAR-T和TCR-T。
在20世纪50年代,移植物抗肿瘤反应的发现以及T细胞在此过程中所起的关键作用,促使人们开始积极探索T细胞在抗肿瘤免疫中的作用。1982年,这一领域的先驱Steven Rosenberg博士首次从多种小鼠肿瘤模型中分离出TIL,随后证明TIL联合IL-2在全部的MC38结肠腺癌肝转移小鼠和一半的肺转移小鼠中产生疗效,这为TIL治疗晚期恶性肿瘤的临床应用点亮了曙光。1988年,TIL首次被应用于临床,在转移性黑色素瘤中的ORR达到了60%。同时,人们也观察到较为矛盾的结果。Gardini在20世纪90年代进行了一项临床试验,研究人员通过从结直肠癌患者的肝转移瘤中提取TIL,在体外用IL-2刺激并扩增,然后将TIL重新回输到患者体内。14例结直肠癌肝转移患者接受了该治疗,遗憾的是,TIL组与传统化疗组的无病生存率(disease free survival,DFS)无显著差异。
目前,许多基于TIL的临床研究已经展开,其中一部分研究已经显示了令人激动的结果,已有自体TIL产品LN-145、LN-144和LN-145-S1用于治疗不能切除或转移的黑色素瘤、复发或转移的头颈部鳞状细胞癌和非小细胞肺癌,目前正处于Ⅱ期临床开发阶段。其中LN-145在27例至少接受过1次化疗的晚期宫颈癌患者中显示出了良好的初步疗效,观察到ORR为44%,包括1例CR和9例PR,FDA已经授予LN-145突破性疗法认定。受此鼓舞,近年来针对TIL的试验数量达到了顶峰,其中黑色素瘤仍然是头号肿瘤类型,其次是非小细胞肺癌、卵巢癌和头颈癌。在CRC领域,2016年,Steven Rosenberg团队在1例转移性CRC患者的肿瘤浸润淋巴细胞中鉴定了针对突变型 KRAS G12D 的多克隆CD8 + T细胞应答,在给患者输注限制性肿瘤浸润淋巴细胞(由四种不同的特异性靶向 KRAS G12D 的T细胞克隆型组成)后,观察到所有7例肺转移病灶的客观缓解。
然而,有几个因素可能会阻碍TIL疗法在CRC患者中的成功应用。由于CRC肿瘤浸润的效应细胞相对较少,因此很难从CRC标本中获得足够数量的TIL。一些研究小组已经致力于通过各种策略在体外有效扩增TIL,同时改进了TIL的分离和增殖技术。另一个问题是肠道内存在菌群,这些菌群经常污染CRC标本,这使得从CRC肿瘤中培养出TIL变得困难。为了绕过这个限制,肿瘤引流淋巴结来源的淋巴细胞(lymph node lymphocyte,LNL)被逐渐开发并进入临床研究。在肿瘤抗原的刺激下,LNL比未受累淋巴结的淋巴细胞显示出更强的增殖潜能,可作为无菌肿瘤特异性淋巴细胞的可靠来源。在20世纪90年代末,Satoh和Triozzi报道了他们通过LNL对转移性结直肠癌患者的研究。通过提取患者的LNL并在体外扩增,他们观察到经过LNL治疗后产生的阳性反应,并且显著延长了患者的生存期。在最近的一项研究中,16例中晚期CRC患者(7例为Ⅱ~Ⅲ期,9例为Ⅳ期)接受了LNL治疗,结果显示所有Ⅳ期患者均有客观缓解,治疗组平均生存期由0.8年显著延长至2.6年,进一步证明LNL在CRC临床应用中的有效性和可行性。基于这些有希望的结果,Jin等对71例CRC患者(46例Ⅰ~Ⅲ期患者接受根治性手术,25例Ⅳ期患者接受姑息性手术)进行了一项大型Ⅰ/Ⅱ期临床研究,试验结果表明,LNL比TIL含有更多活化的CD3 + CD69 + 和CD4 + CD69 + T细胞。与对照组相比,LNL组患者的OS显著改善(28个月vs. 14个月),且未观察到任何副作用。TIL疗法是一种高度个性化的疗法,效应细胞是经过天然选择与富集、肿瘤特异性T细胞比例高、多样性丰富的群体。与其他免疫疗法相比,TIL具有多靶点、肿瘤趋向和浸润能力强、副作用小等优势,是实体瘤治疗的理想选择,前景广阔。除此之外,在成本方面,有业内专家预计,TIL疗法的价格为7万~8万美元,低于目前CAR-T细胞治疗数十万美元的治疗费用,具有一定的经济优势。
在过去的几年里,针对细胞免疫治疗开展的多中心临床试验数量日益增长,其中CAR-T细胞治疗是一个有前途的肿瘤治疗手段。然而,不同于CAR-T细胞治疗在血液系统肿瘤中的光明前景,CAR-T细胞治疗实体肿瘤仍然处于较为落后的境地。尽管如此,已有多项针对CRC CAR-T细胞治疗的临床试验,表9-2-2总结了临床试验的详细信息。大多数试验处于临床Ⅰ期或Ⅱ期,主要研究目标是评估安全性、剂量水平和最大耐受剂量。
表9-2-2 结直肠癌患者CAR-T细胞治疗的临床试验
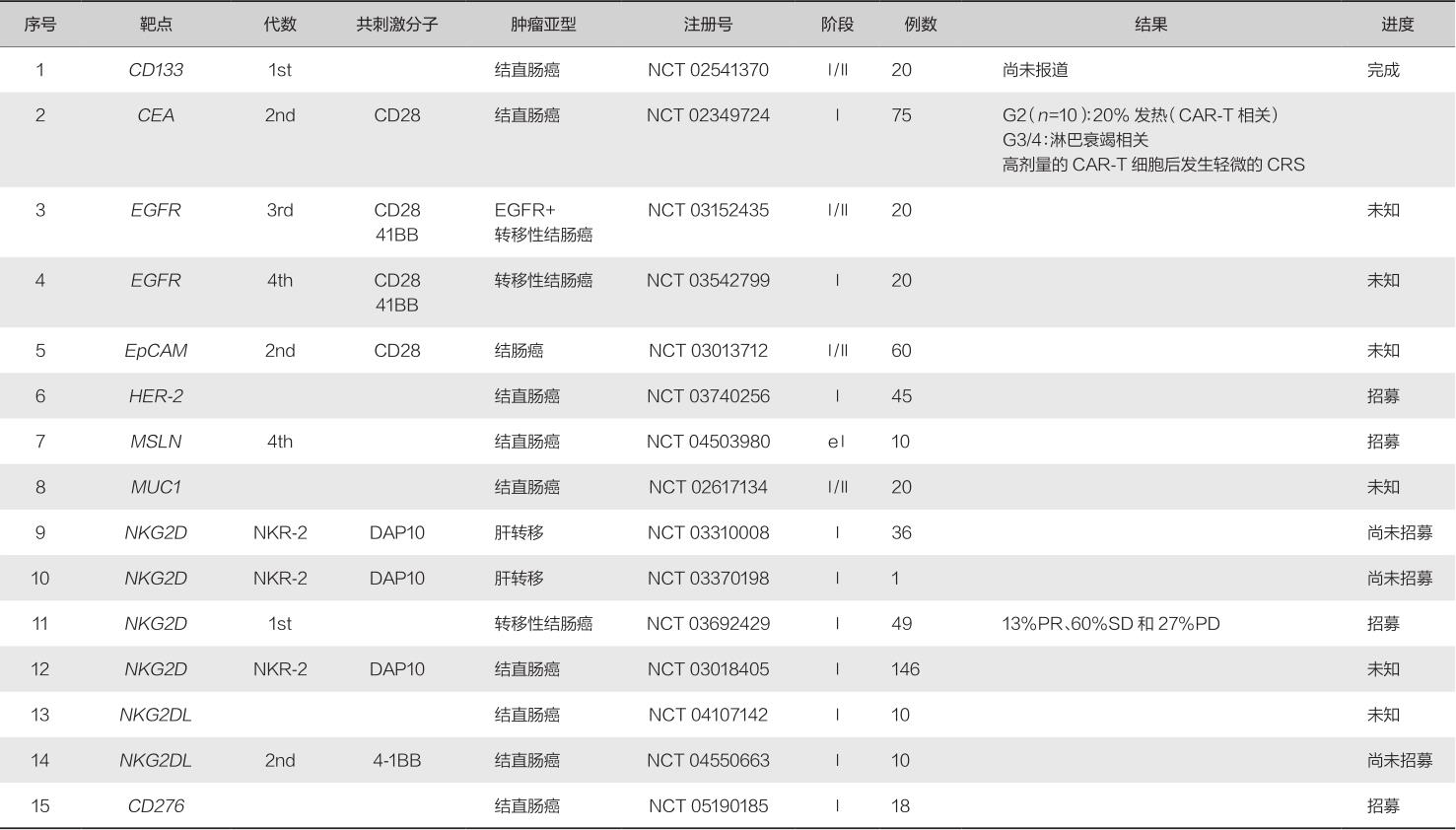
注:PR.部分缓解;SD.病情稳定;PD.进展性疾病。
CRC CAR-T细胞治疗中研究最多的靶点是 CEA 和 NKG2DL ,其次是 EGFR 和 HER - 2 。第一个CRC的CAR-T临床试验开始于2014年,探索了第二代CEACAR-T细胞在CRC患者中的安全性和有效性,也包括肺癌、胃癌、乳腺癌和胰腺癌患者(NCT 02349724)。在接受CAR-T细胞治疗后,10例既往治疗的进展性疾病患者中有7例病情稳定(stable disease,SD),2例SD持续超过30周。此外,大多数患者血清CEA水平下降,2例患者出现肿瘤缩小。2例患者在CAR-T细胞治疗后的几个小时内经历了发热,并且在细胞输注后的1~4天多次发热,根据需要给予非甾体抗炎药治疗。同样类型的抗CEA-CAR-T细胞还在CEA阳性结直肠癌伴肝转移患者中进行了研究(NCT 02416466和NCT 02850536)。前者通过肝动脉灌注来改善CAR-T细胞向肝转移病灶的转运,并且直接经动脉肝灌注有助于降低发生CRS的风险,这是一种在CAR-T细胞全身注射方案中常见的威胁生命的并发症。第二个试验通过产生有利的压力来克服肿瘤微环境的输注屏障,将CAR-T细胞传递到肝转移瘤的效率提高了5.2倍。
除了经典靶点,一个由NKG2D细胞外区域作为配体结合区域和内源性DAP10作为共刺激区域组成的非典型CAR已被用于3个不同的研究。尽管临床试验尚未报道疗效结果,但NCT 03018405试验的初步结果表明,每剂量CAR-T细胞展现出良好的安全性。鸟苷酸环化酶C(guanylyl cyclase C,GCC/GUCY2C)属于受体鸟苷酸环化酶家族中的一员,在胃肠道的液体和离子稳态中起关键作用。近年来研究发现,GCC在原发性结直肠癌细胞中稳定表达,而在转移性结直肠癌细胞中异常高表达,被认为是转移性结直肠癌的特异性标志分子之一。因此研究人员开发了GCC19-CAR-T,作为一款自体CAR-T治疗产品,被用于治疗复发难治转移型CRC。公开资料显示,此前GCC19-CAR-T在中国进行的针对复发难治型CRC患者的临床试验已取得初步成果。研究者累计入组35例晚期CRC患者,在剂量爬坡试验中接受了2×10 6 /kg的GCC19-CAR-T细胞的8例患者中,观察到ORR为50%。2022年4月19日,我国开发的实体瘤CAR-T产品GCC19-CAR-T被FDA授予快速通道资格。
多数临床试验使用的是第二代CAR,但实体瘤的微环境包含丰富的纤维基质和免疫抑制细胞,保护肿瘤组织并抵抗免疫细胞攻击。最新的第四代CAR进入临床研究,有望改善这一现状。Tian开展了能够诱导产生和释放IL-12细胞因子EGFR-CAR-T细胞治疗结直肠的临床试验(NCT 03542799),它能够调节肿瘤微环境,吸引和激活先天免疫细胞攻击肿瘤。另一种第四代CAR是基于MSLN-CAR-T,同时能够诱导产生和分泌抗PD-1的纳米体(NCT 04503980),增加免疫细胞的应答。此外,目前临床试验中有两种针对CRC的同种异体基因CAR-T细胞治疗(NCT 04107142和NCT 03692429)分别于2019年和2020年启动。两种CAR-T细胞治疗都是针对NKG2D配体,但不同的策略使它们适合异体使用。
T细胞受体修饰是另一种ACT,被称为TCR转导疗法,与CAR-T细胞非常相似,但它们识别抗原的机制却完全不同。在CAR-T细胞中,抗体片段用于结合癌细胞表面的特异性抗原。然而,TCR使用由α和β肽链组成的异二聚体识别MHC分子呈现的多肽片段,不仅能够识别突变后肿瘤细胞产生的细胞表面抗原,还可以识别细胞内抗原或新抗原。第一份关于结肠癌TCR-T细胞治疗的研究报告以CEA抗原为靶点,发现了一些临床反应的证据,但由于结肠正常细胞中也存在CEA,因此出现了严重结肠炎。这既证明了T细胞治疗转移性结肠癌的可行性,也证明了将CEA作为抗原靶向的局限性。继有希望的结果之后,另一种尝试靶向转化生长因子β受体Ⅱ型(TGF-βRⅡ)移码抗原(MSI-H结肠癌中表达)的mRNA工程化T细胞被开发用于治疗晚期转移性结肠癌患者。然而,由于严重的不良反应,临床试验终止。其他正在进行的TCR治疗针对 KRAS G12V+ 肿瘤(NCT 03190941)和 KRAS G2D+ 肿瘤(NCT 03745326),两者都处于临床试验Ⅰ期/Ⅱ期,期待在不久的将来带来治疗肿瘤患者的新武器。
T细胞受体治疗虽然是一种非常有前途的方法,但也面临许多挑战,包括良好的靶点选择、寻找特异性TCR、最佳TCR亲和性筛选、安全性评估等。此外,由于TCR治疗高度依赖MHC进行抗原提呈,考虑到肿瘤环境中MHC分子的下调或突变,带来一定的临床应用局限性。简而言之,TCR-T细胞治疗已显示出一定的治疗潜力,但仍有许多局限性需要仔细考虑。
其他的ACT还包括自体淋巴因子激活的杀伤细胞、CIK和DC-CIK。但是相关研究较为零散,病例数有限,往往难以做到严格的双盲和随机,其临床价值仍需要更多的临床试验来验证。曹雪涛教授团队开展的Ⅱ期随机对照临床研究对既往的DC-CIK治疗进行改进,将患者自身肿瘤抗原负载DC细胞,进而能更有效地诱导机体特异性免疫应答,并联合mFOLFOX(奥沙利铂+氟尿嘧啶+亚叶酸钙)化疗方案治疗晚期CRC。主要终点ORR获得阳性结果,初步数据显示,DC+CIK联合化疗组疾病缓解率达45.1%(32/71),而单纯化疗组为25.4%(18/71)。细胞因子治疗在肿瘤微环境中增强局部抗肿瘤免疫反应的同时,也会诱导免疫系统的广泛激活,进而诱发治疗相关的毒性反应,甚至导致死亡。因此近年关于CRC单一细胞因子治疗的研究非常有限。
过继细胞疗法的兴起为晚期难治性肿瘤患者带来了新的希望,但是其制备过程相对复杂,耗时长、花费大,细胞表面缺乏与实体瘤分泌的趋化因子相匹配的受体,造成CAR-T对肿瘤部位的归巢能力差,存在脱靶效应,并且严重的细胞因子风暴也会给患者的治疗带来极大风险,所以ACT在临床的广泛应用还有很长的路要走。
首先,ACT的研究在未来应该着重解决其安全性及有效性问题。提高过继细胞激活程度、提高其增殖能力、延缓细胞衰竭、延长细胞在患者体内的存活时间是未来研究的重点方向之一,可以基于特定肿瘤微环境选择合适靶点,或者联合免疫抑制剂等其他治疗,解除肿瘤微环境的免疫抑制作用,增强CAR-T细胞的浸润和杀伤能力,提升CAR-T细胞的有效性;也可开发一些控制程序来调控细胞活性,通过安装自杀开关对移植细胞进行快速清除,降低细胞毒性,提高治疗安全性;同时可以利用合成生物学方法,优化CAR-T细胞制造,改善CAR-T细胞的代谢适应性,探索预防或逆转T细胞耗竭的新方法。
其次,由于现在使用的CAR-T细胞都来源于自体血液,这使得当前的CAR-T细胞疗法高度个性化。并且由于自体CAR-T细胞的采集耗时久,部分患者的免疫细胞状态无法达到制作CAR-T的标准,导致自体CAR-T细胞疗法的应用受到一定限制。所以通用型CAR-T的开发也是重要的研究方向。该技术主要依赖于基因编辑平台,使用CRISPR技术敲除T细胞中引起免疫排斥的基因,使其成为能够表达靶向癌细胞的CAR-T细胞。通用型CAR-T的上市将会大大降低CAR-T疗法的成本,使这项技术惠及更多的患者。
最后,ACT领域不会孤立发展,最好的肿瘤响应和维持需要ACT结合免疫治疗及化疗等多种治疗的协同作用。相信随着科技的发展,ACT将为肿瘤患者带来新的希望。
肿瘤新抗原(neoantigen)的概念在20世纪90年代就被提出,为肿瘤细胞中存在的突变基因编码异常的蛋白。该异常的蛋白被降解为肽段之后,肽段被细胞的抗原提呈系统(MHC I)识别,提呈到肿瘤细胞表面,成为肿瘤细胞表面特异性的标志物,理论上体内的免疫系统可以通过识别这些新抗原对肿瘤细胞进行特异性的杀伤。与肿瘤相关抗原(tumor-associated antigen,TAA)和癌-睾丸抗原(cancer-testis antigen,CTA)不同,这些能够激活免疫系统的突变蛋白只表达于肿瘤细胞,称为肿瘤特异性抗原(tumor-specific antigen,TSA);同时由于是肿瘤细胞新产生的抗原,故而被称为新抗原。
新抗原也可以通过病毒感染、选择性剪接和基因重排产生。新抗原为肿瘤细胞所特有,正常细胞中几乎不表达,可区分肿瘤细胞和正常细胞,常具有强免疫原性,因此针对新抗原的治疗不易发生脱靶现象;此外,由于新抗原未经过中枢免疫耐受过程,相较TAA具有更强的免疫原性,这些特点使得新抗原成为肿瘤免疫治疗的优秀靶点。
肿瘤新抗原可分为两类:共享新抗原和个性化新抗原。共享新抗原是指在不同癌症患者中常见的、不存在于正常基因组中的突变抗原。具有高度免疫原性的共享新抗原,有可能被筛选为具有相同突变基因患者的广谱治疗性癌症疫苗。个性化新抗原是指大多数新抗原所特有的、因患者而完全不同的突变抗原。因此,个性化新抗原制剂药物只能特异性地针对每个患者,即个性化治疗。
驱动基因突变的频率表明,靶向其共享新抗原可能会使大部分患者受益。除癌基因外,一些抑癌基因也表现出复发突变,但一般抑癌基因不太可能是共享新抗原的来源,因为它们经常被非复发突变灭活或由于mRNA转录物无义介导的衰变而低水平表达。与个性化新抗原相比,共享新抗原靶向免疫治疗也有利于共享相同新抗原的患者。例如,新抗原预测在早期进行,通常针对每个患者个性化。但只要在患者的肿瘤细胞中检测到这样一种共享新抗原,就可以直接应用到相应的现成新抗原靶向免疫疗法中进行验证和治疗,可以大大缩短研发周期。
新抗原具有较强的免疫原性,可降低肿瘤细胞免疫逃逸的概率。然而,同一肿瘤不同个体中新抗原种类和数量的不同,表现出明显的个体异质性。针对共享新抗原的现成精确免疫治疗将广泛适用于许多患者,并在可扩展性方面相对于个性化新抗原具有巨大优势。但现成的共享新抗原治疗的一个潜在缺点是,没有一个患者可能有一个以上共享新抗原可供靶向。癌症通常有多个个性化新抗原,原则上有助于组合靶向,并降低由于克隆进化过程中抗原丢失而产生治疗抵抗的风险。
因此,新抗原在肿瘤免疫治疗中的应用将趋于个性化。个体化的癌症疫苗可以单独或与其他疗法联合作用,以增加强度和持久的抗肿瘤作用,提高患者的生存率和生活质量,最终改善癌症患者的治疗结果。个体化癌症疫苗治疗癌症患者的可行性、安全性和免疫原性决定了它将是未来重要的发展趋势。预计在可预见的未来,个体化的癌症疫苗将使大多数患者获得精准治疗。
目前新抗原靶向免疫治疗的框架包括4个主要步骤。首先,通过全外显体测序从患者的肿瘤和正常组织中识别体细胞突变。其次,通过预测主要组织相容性复合体肽结合亲和力的生物信息学算法,预测和优先考虑最有吸引力的新抗原作为免疫治疗的靶点。再次,验证所选新抗原的免疫原性和肿瘤反应性。最后,为选定的新抗原开发个性化的免疫疗法,如个性化的疫苗接种或采用T细胞疗法。
虽然新抗原在肿瘤治疗方面取得了良好的临床进展,但具有免疫原性的新抗原数量较少,且预测比较困难。因此,新抗原领域需要更多优化的算法和验证方法进行准确的预测,以选择更可靠的高免疫原性新表位。目前,肿瘤新抗原的预测准确性仍然是一个亟待解决的问题。肿瘤新抗原预测算法需要考虑很多因素,包括HLA分型、表达、突变分析、预测肽处理、TCR结合力、MHC亲和力、抗原肽-MHC分子复合物(peptide-MHC,PMHC)稳定性、肿瘤新抗原来源等。此外,还包括T细胞识别、TCR分析和免疫细胞分析,以评估T细胞反应。对于新抗原筛选和T细胞反应评估,除了二代测序,还有高分辨率和串联质谱技术以及用于肽预测的生物信息学技术,但基于机器学习和AI技术的预测算法需要使用验证性数据集持续训练,其中数据类型、质量和数量会极大地影响算法精度。因此,数据库的不断积累,特别是已验证的肿瘤新抗原数据,对于提高算法的准确性是极其关键的。肿瘤新抗原筛选联盟是由帕克癌症免疫治疗研究所(Parker Institute of Cancer Immunotherapy,PICI)和癌症研究所(Cancer Research Institute,CRI)发起并形成的。该联盟的目标是为全球新抗原检测建立算法和标准,共同努力预测更精确的抗癌靶点,推进个性化肿瘤疫苗的研究和应用。
用于新抗原鉴定的常用筛选方法可以分为3种。方法一,设计成熟的新肽疫苗疗法。第一步是识别肿瘤特异性突变,与免疫识别相关的突变主要包括具有外显子、内嵌子和融合基因的非同义单核苷酸变异体。配对肿瘤和正常细胞DNA的全外显体测序是识别体细胞突变的最常用方法。通过RNA测序分析鉴定、突变、正交验证和等位基因的表达水平,然后根据预测的与自体HLA Ⅰ类和Ⅱ类的高亲和力对突变进行排序。免疫表位数据库和分析资源是一个由T细胞表位和工具组成的在线综合数据库,可以用来预测MHC结合。免疫表位数据库和分析资源提供的预测工具包括基于人工神经网络(artificial neural network,ANN)的深度学习模型NETMHC、NETMHCPAN、SMM、SMMPMBEC、ARB、COMBLIB_SIDNEY2008、Pickpocket和Consulnal。合成的新肽的免疫原性必须通过T细胞反应性分析来验证,通过产生抗原负载的自体抗原提呈细胞来刺激T细胞。然后必须检测CD4 + 和CD8 + T细胞的激活标记,包括OX-40、4-1BB、CD170A和IFN-γ。2014年初,小鼠疫苗接种证实了上述途径。
从方法一简化,在挖掘非同义单核苷酸变异体之后,串联合成编码突变的多个微基因,以便在方法二中生成串联微基因(tandem microgene,TMG)构建体。TMG构建体由基因融合在一起的可变数量的微基因组成,每个微基因编码来自内源性蛋白质序列的12个氨基酸两侧的突变。利用编码TMG构建体的质粒作为模板产生体外转录RNA。2014年,Rosenberg团队利用这种方法从一名转移性胆管癌患者中识别出一个ErbB2结合蛋白质(ErbB2 interacting protein,ErbB2IP)突变。再过继转移TIL,其中含有约25%的突变特异性T细胞,患者经历了肿瘤消退。在连续的研究中,他们继续证明10例转移性胃肠道癌患者中有9例的新表位可以被自体TIL识别。
方法三基于数据库和文献识别新表位,无须样本获取。这一策略的关键是高频突变位点的存在。基于这种模式,舒马赫等在弥漫性Ⅱ级和Ⅲ级胶质瘤患者中发现了最常见的突变 IDH1 (R132H),随后合成了一种针对突变 IDH1 的肽疫苗,该疫苗的功能是诱导小鼠的抗肿瘤反应。同样,Platten等发现 K27M 突变的Histone-3作为胶质瘤疫苗产生的最佳靶点,证明针对 K27M 突变的Histone-3的肽疫苗在MHC人源化小鼠模型中引发突变特异性免疫应答。理论上,其他高频突变,包括 BRAF 、 RGFR 和 KRAS 也可以作为理想的癌症疫苗靶点。
近年来,基于新抗原的CRC研究获得了很多进展。Bakarurraini等通过癌症基因组图谱(TCGA)队列,利用肿瘤特异性新抗原数据库(TSNAdb)来预测CRC中的新抗原。鉴于 TTN 基因是一个大基因,因此它在CRC中拥有最高数量的预测新抗原。同样, TTN 基因也是乳腺癌新抗原数量最多的基因之一。 TTN 编码一种结构蛋白,尽管其与癌症的生物学作用仍有争议,但一些研究表明 TTN 基因与免疫学相关。例如,有研究表明, TTN 突变能够预测不同实体肿瘤免疫检查点阻断后的预后。基于抗原的研究,Parkhurst等的研究表明,突变的TTN肽能够诱导CD8 + T细胞反应,但与其他受试抗原相比,其诱导率较低。除 TTN 外, MUC16 基因也被预测会产生大量新抗原。除CRC外,其他癌症也被预测会产生更多针对 MUC16 基因的新抗原,如胃癌和胰腺癌。研究发现,MUC16新抗原能够在胰腺癌长期存活者中引发CD8 + T细胞反应。该研究使用了来自 MUC16 基因的两种新肽:MGKSTHTSM和VMKHLLSP肽。
Mennonna等的一项研究,通过高通量测序在CRC样本中发现了新的体细胞突变。除了在CRC中发现的常见突变基因,研究人员还在 Smad4 基因中发现了新的突变,发现 Smad4 V370A表位在体内具有免疫原性,最重要的是,在携带这种突变的患者自体T细胞中发现了针对这种特异性突变的循环T细胞。Newey等分析验证了612个非沉默突变中的三个新抗原,它们是来自 U2SURP 、 MED25 和 FMO5 基因的肽,产生的肽长度为8~11个氨基酸。
CRC中移码/插入/缺失突变衍生的新抗原ICI,如抗CTLA-4、抗PD1/PDL1,作为癌症治疗方案的一部分,已使多种癌症的预后显著改善,特别是黑色素瘤和非小细胞肺癌。然而,这种治疗和化疗的疗效仅限于某些癌症类型。CRC与MSS和pMMR卵巢癌患者从该治疗中获益不多。
据报道,低错配修复率(MMR)和微卫星不稳定(MSI)的CRC患者在ICI治疗时表现出比MSS和pMMR患者更好的预后。这一发现强调了利用患者染色体不稳定性的因素作为一种有效治疗的新方法。高突变负荷的肿瘤存在dMMR机制缺陷,导致高MSI。此外,ICI治疗MSI-H CRC的疗效优于MSS CRC。由于观察到这种类型的CRC有较好的预后,基因组中的移码突变可能导致肿瘤特异性新抗原的合成,这些新抗原能够诱导抗肿瘤反应。据Ozcan等报道,大多数MMR缺陷的癌症会引起干扰HLA Ⅰ类抗原提呈的突变,反映了肿瘤发展过程中的免疫监视和主动免疫选择。此外,MSI/dMMR肿瘤与高TMB和高免疫原性新抗原有关,这些新抗原来自移码突变。正如Maby等所证明的,移码突变与较高的肿瘤特异性免疫和肿瘤浸润性或/和新抗原特异性CD8 + T细胞密度之间存在相关性。CRC组织的MSI状态最终影响突变的数量,进而影响可能的新抗原数量。根据TCGA数据集,如从癌症免疫图谱中获得的,MSI的CRC比MSS组有明显更多的突变和预测的新抗原。
Rospo等在一组CRC细胞系中也表明了这一点,MSI细胞系比MSS细胞系和携带极点基因突变的细胞系具有更高的负荷或突变负荷。在MSI肿瘤中,TIL的百分比高于MSS肿瘤,这是由于移码突变的增加。这些移码突变可能产生免疫原性移码突变衍生的新抗原,可以吸引更高的CD8 + T细胞密度。Tougeron等的一项研究显示,MSI CRC肿瘤中 ACVR2 、 TAF1B 、 ASTE1/HT001 和 TGFBR2 基因突变率最高。此外,他们还表明TIL的最高密度与 ASTE1/HT001 和 PTEN 的突变有关,这与Maby等的研究一致,表明当 ASTE1 、 HNF1A 或 TCF7L2 基因发生突变时,TIL密度增加。本研究还表明,MSI CRC具有不同的免疫微环境,由肿瘤新抗原特异性CD8 + T细胞组成,可用于进一步推进该特异性肿瘤亚群的免疫治疗,包括林奇综合征。
在过去的几十年里,移码衍生肽(frameshift derived peptide,FSP)在MSI CRC中的研究一直在进行。最早的研究之一是 TGFBR2 基因在MSI CRC中的表达。 TGFBR2 在90%左右的CRC组织中发生突变,并经常导致缺失或插入。研究人员产生了几种对应基因突变区域的肽,并进行了基于T细胞的检测,发现P538肽具有最高的免疫潜能。TIL对P538的反应能力以及MSI患者T记忆细胞的产生证明了这一点。在同一项研究中,研究人员还对Bax衍生的突变肽进行测试,但没有观察到阳性结果。随后,同一组能够进行进一步的体外试验,并显示当用相同的TGFBR2肽脉冲的抗原提呈细胞重复刺激T细胞时,T细胞能够杀死含有突变肽的细胞系。另一项研究以 TGFBR2 突变体中的3种不同的多肽为研究对象,成功地证明了T细胞诱导的FSP02多肽能够以HLA-A201依赖的方式裂解携带该突变体的细胞系。此外,从TGFBR2衍生的FSP的免疫原性也在体内模型和随后的临床试验中得到证实。
除TGFBR2外,还对来自突变基因的其他FSP进行了检测,如 OGT 、 HPDMPK 、 D070 、 U79260 、 FLT3L 等。同样的研究表明来自 OGT 和 U79260 的FSP也具有免疫原性。有趣的是,同组用不同的细胞系(阳性携带突变和HLA-A201)重复研究来自 OGT 的FSP,用相同肽脉冲的CTL能够裂解携带 OGT 突变蛋白的细胞系。除此之外,这个小组还鉴定了其他几个来自不同基因的FSP,如 caspase 5 、 taf - 1b 和 ht001 。在检测的5个FSP中,研究者从 caspase 5 中鉴定出一个FSP,其肽序列为FLIIWQNTM,能够以HLA-A201方式诱导CTL介导的裂解。此外,还鉴定了 MSH03 基因的免疫原性FSP。Garbe等测试了12个不同的FSP,并从 MSH03 基因中鉴定了两个HLA-A0201限制性细胞毒T细胞表位。此外,还发现了一个新的HLA-A0201限制性细胞毒T细胞表位,该表位来自 U79260 (FTO)突变区。Speetjen等进行的一项较大规模的研究表明,使用移码突变抗原在MSI癌中常见,包括CRC、胃癌和子宫内膜癌。该研究从 TGF - R2 - 1 、 MARCKS - 1 、 MARCKS - 2 、 CDX2 - 2 、 TAF1B - 1 、 PCNXL2 - 2 、 TCF7L2 - 2 和 Bax α- 1 等不同基因中鉴定了8个可能感兴趣的移码突变抗原。
迄今为止,大多数基于新抗原的研究都集中在错义突变,以识别免疫原性表位。融合基因提供了一个吸引人的新抗原来源,特别是框架外基因融合。这是因为新的帧外序列的存在可能会产生一个新的开放阅读框架,并随后产生一个可能具有免疫原性的新肽的长序列。Rasthe等最近的一项研究已从骨肉瘤中鉴定出融合基因衍生的新抗原,本研究优化了一条生物信息学系统,以识别具有潜在新表位的融合转录本。在Yang等的另一项研究中,他们鉴定了DEK-AFF2融合衍生肽,该肽在低肿瘤突变负担的头颈部癌中引发T细胞反应。在CRC方面,MSS CRC携带的融合基因比MSI CRC高,胃癌和子宫内膜癌也有相同的表达模式。此外,来自融合基因的候选新抗原比SNV和INDEL衍生的候选新抗原具有更强的免疫原性。值得注意的是,在TCGA队列中,具有最高免疫原性潜力的候选新抗原来自基因融合,从而使它们成为癌症疫苗的更好候选。
多项研究表明,由SNV引起的常见突变也能够产生可检测的新肽,并引发特异性免疫反应。例如,Illzumi等最近的一项研究揭示了一些常见突变基因,如 KRAS G12 和 KRAS G13 能够刺激健康人外周血单核细胞PBMC的免疫应答。结果表明,所合成的多肽能够诱导人白细胞抗原(human leukocyte antigen,HLA)Ⅱ类限制性CD4 + T细胞应答。有研究显示,在1 779例CRC患者的全外显子组测序中发现了复发的新抗原,在5例患者中发现1 550个突变,其中 KRAS G12V (5.8%)、 KRAS G12D (8%)、 PIK3CAE545K (3.5%)、 BMPR2N583TFS44 (2.8%)和 PIK3CAH1047R (2.5%)在转移性泛癌中的突变率较高,可能是肿瘤免疫治疗的靶点。 KRAS 突变是一个主要的突变,该突变与CRC的Th1细胞毒免疫抑制有关,为CRC的生物学异质性增加了一个新的免疫生物学方面。同样,Veatch等发现部分非小细胞肺癌患者的CD4 + T细胞能够识别 KRAS G12V 突变肽而不是野生型肽,这与Quandt等的另一项研究一致,在该研究中,他们产生了由 KRAS 和 TP53 中常见突变组成的长肽,并发现了针对携带这些突变的胃肠道肿瘤的更高效应T细胞。Lo等报道了对识别突变 P53 p.R175H 的T细胞受体(TCR)的鉴定和表征,该突变在癌症患者亚群中共享,他们对一例转移性结直肠癌患者的肿瘤浸润淋巴细胞进行筛查,以识别突变的新抗原,发现HLA-A*0201-限制性识别突变 P53 p.R175H ,最小肽表位为HMTEVVRHC。四聚体分选分离反应性T细胞,鉴定出3个TCR。这些TCR介导识别卵巢癌、子宫癌和骨髓瘤细胞系,以及NIH患者来源的食管腺癌细胞系,该细胞系内源性表达 P53 p.R175H 和 HLA - A*0201 ,再用编码 HLAA*0201 的逆转录病毒转导后,他们还介导了 P53 p.R175H 结肠、乳腺和白血病细胞系的识别。这项工作表明,共同的共享突变表位,如在P53中发现的那些,可以引起免疫原性反应,并且ACT的应用可以扩展到任何组织学上同时表达 HLA - A*0201 和 P53 p.R175H 突变的癌症患者。这些研究表明,常见的突变可能比最初认为的更具免疫原性,这为更广泛的人群开辟了一条普遍靶向治疗的新途径。
肿瘤疫苗,即肿瘤特异性主动免疫治疗,是20世纪90年代发展起来的肿瘤免疫新疗法。其基本原理是:通过体外分离、提取TSA或TAA,制备不同形式的疫苗注射到肿瘤或肿瘤患者体内,由抗原提呈细胞摄取并提呈给免疫细胞,使机体T细胞致敏、活化,生成肿瘤特异性细胞毒性T细胞,转移性地结合并杀伤肿瘤细胞。理想中的肿瘤疫苗不仅需活化刺激CD8 + T细胞,产生肿瘤特异性细胞毒性T细胞,而且还要能够活化刺激CD4 + 辅助性T细胞,并产生功效,最终产生强有力的、持续有效的抗肿瘤免疫效应。
近年来大力发展的肿瘤新抗原疫苗是高度个体化疫苗,相对安全且副作用小,在CRC临床研究中展现出了积极的疗效。随着疫苗技术的不断完善,其他的传统肿瘤疫苗类型,如生物大分子疫苗、多肽疫苗、肿瘤细胞疫苗等,也重新引起重视并取得了一定发展。
在体内已观察到诱导有效的新抗原特异性抗肿瘤T细胞免疫应答和抑制肿瘤生长的现象。这些临床前试验突出了新抗原免疫疗法作为一种新的治疗策略的潜力。新抗原疫苗的有效性也在CRC小鼠模型中得到证实。新抗原疫苗已用于黑色素瘤和胶质母细胞瘤的临床试验,证明疫苗是一种诱导肿瘤特异性T细胞应答的安全方法。此外,目前有一些临床试验探索不同类型新抗原疫苗对CRC患者的有效性和安全性。
一项针对 KRAS 突变肽的初步研究表明,在7例CRC患者中,只有2例在接种疫苗后出现阳性免疫反应。Kloor等最近进行了一项Ⅰ期临床试验,评估移码肽新抗原疫苗对dMMR CRC患者的安全性和免疫原性。该试验使用了基于 AIM2 、 HT001 和 TAF1B 突变引起的FSP新抗原的疫苗,结果显示,所有22例dMMR CRC患者中至少有一种移码肽疫苗诱导了体液和细胞免疫反应,没有出现严重的疫苗相关不良反应。
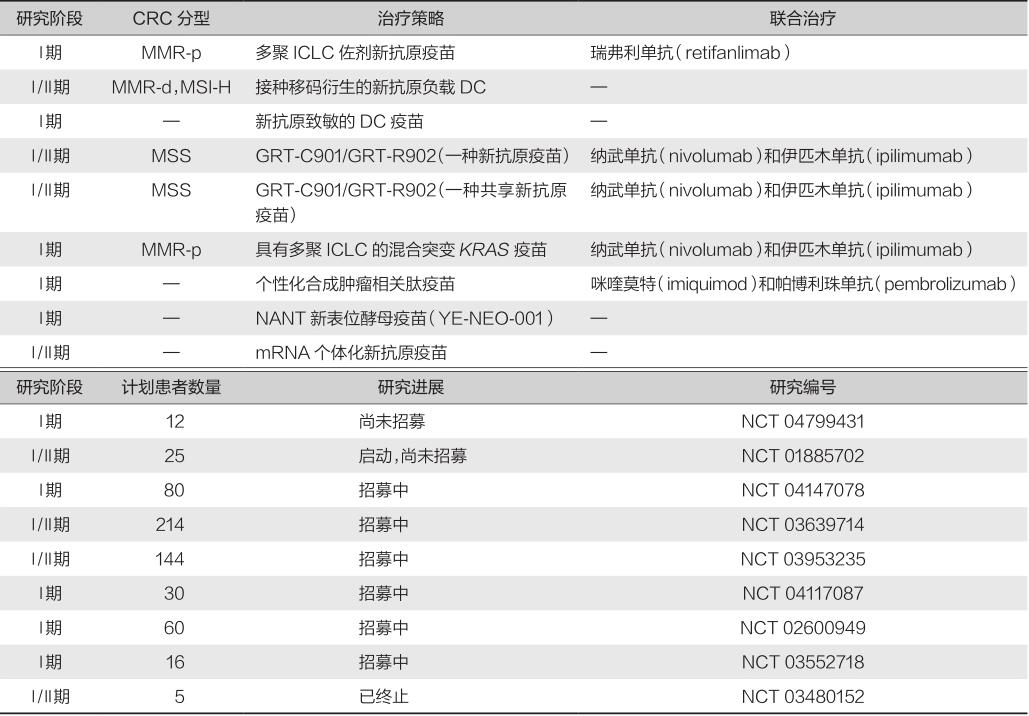
通过使用GM-CFS作为佐剂来增强对新抗原疫苗的免疫反应的尝试获得了成功。然而,未发现该疫苗对患者疾病进展有利,这可能与免疫调节细胞的增加有关。此外,许多新抗原疫苗已在临床前小鼠模型中进行了试验。Ni等开发了一种新抗原(Adpgk)纳米疫苗(banNV),使用Toll样受体7/8激动剂R848和TLR9激动剂CpG作为双佐剂。研究发现,双佐剂新抗原疫苗增加了新抗原的免疫原性,并与抗PD-1治疗一起产生了良好的抗肿瘤反应。也有报道称,使用多种新抗原DNA疫苗和抗PD-1治疗可协同控制MC38结肠癌细胞系的生长。Kim等报道,基于新抗原的EpiGVAX疫苗与5-aza-2′-脱氧胞苷联合使用,可通过诱导新抗原特异性抗肿瘤T细胞应答,提高辐照后的全细胞CRC疫苗的抗肿瘤效果。Leoni等最近从MSI CRC基因组图谱数据库中选择了209个共享FSP来生产病毒载体疫苗nus-209,通过体外实验证实该疫苗可以激活人类CD8 + T细胞。表9-2-3总结了结肠癌领域开展的肿瘤新抗原疫苗临床研究汇总。
表9-2-3 结肠癌领域开展的肿瘤新抗原疫苗临床研究汇总
树突状细胞(dendritic cell,DC)通过白细胞分离得到,是人体内最有效的抗原提呈细胞之一。DC在细胞因子的培养下成熟,并与外源性肽或肿瘤裂解液混匀,最后输注给患者。DC疫苗在黑色素瘤和前列腺癌的临床试验中显示出良好效果,在CRC中也进行了一系列探索。Rodriguezet等进行了一项随机对照试验,纳入了19例可接受手术的结直肠癌肝转移患者。其中15例患者在接受术后辅助化疗后随机分为接种DC疫苗组和观察组。DC疫苗组和观察组的平均DFS分别为25.26个月和9.53个月(95%置信区间5.32~18.88)。MelCancerVac是一种由DC组成的疫苗,由DDM-1.13的异基因黑色素瘤细胞溶解液混合产生,其MAGE-A3高表达,而MAGE-A3也是CRC中过表达的TAA。一项Ⅱ期试验纳入了20例Ⅳ期CRC患者,每两周接受10次皮内接种。尽管该试验的总体结果没有显示出OS有很大改善(中位OS7.4个月),但5例患者PFS超过6个月,其中2例分别超过27个月和37个月后疾病仍未进展。基于前期数据,计划对CRC患者进行进一步的MelCancerVac Ⅲ期临床试验,后续结果值得进一步关注。
基于生物大分子的疫苗包括多肽/全长蛋白疫苗、DNA疫苗和RNA疫苗。蛋白疫苗一般常包含有丰富的免疫原性表位(TAA或TSA),可由MHC Ⅰ/Ⅱ类分子处理和提呈。在一项纳入96例晚期CRC患者的Ⅱ期临床研究中,5种HLA-A*2404限制性肽(RNF43、KOC1、TOMM34、VEGFR 1、VEGFR 2)混合接种与基于奥沙利铂的化疗方案联合应用被证明是安全的。另一项Ⅰ期临床研究表明,KOC1/TTK/URLC10/DEPDC1/MPHOSPH1联合制备的HLA-A*2404限制性疫苗是安全有效的,入组患者平均总生存期可达9.4个月。
DNA疫苗是通过质粒将编码肿瘤抗原的基因序列引入体内,然后,MHC Ⅰ/Ⅱ类分子提呈转录和翻译的产物。此外,DNA结构还可以通过细胞质传感器激活固有免疫。然而,目前尚缺乏稳定的制造技术来生产可以运输到核膜中的疫苗。此外,由于质粒可以整合到宿主基因组中,这在一定程度上增加了产物的不确定性。
RNA疫苗是将编码TSA的外源基因导入个体内,通过免疫系统合成TSA蛋白,诱导机体产生免疫应答。mRNA疫苗是在体外合成RNA编码的肿瘤抗原,mRNA疫苗进入靶细胞后,可在细胞质内完成翻译而不进入核膜。与DNA疫苗相比,mRNA疫苗更有效,更容易进行个性化的修饰,其生产过程也更为方便。mRNA-4157是一种基于mRNA的肿瘤疫苗,在Ⅰ期临床试验中与PD-1单抗联合给药时表现出良好的安全性和临床效率(NCT 03313778)。近期,一项针对 KRAS 阳性肿瘤的疫苗mRNA-5671联合PD-1单抗治疗微卫星稳定(MSS)肿瘤患者的Ⅰ期临床试验正在启动进行中(NCT 03948763),结果尚未公布。
肿瘤细胞疫苗是一种利用整个肿瘤细胞或裂解物来启动免疫系统的方法。根据肿瘤细胞的来源,肿瘤细胞疫苗可分为自体细胞疫苗和异体细胞疫苗。其中,自体细胞疫苗对于个体的特异性更强,而异体细胞疫苗生产更方便,从而有利于大规模群体应用。由于未知抗原的大量存在,减少了免疫耐受的可能性。然而,由于肿瘤细胞疫苗还含有部分在正常组织中广泛表达的抗原,可能会引起一定的自身免疫反应。
OncoVax是20世纪80年代早期临床试验中研究开展最广泛的CRC疫苗,由一种自体癌细胞与芽孢杆菌Calmette-Gu é rin(BCG)疫苗组合,先后开展了数项研究证实了这种疫苗作为术后辅助治疗手段的显著效果。ECOG-5383研究是一项Ⅲ期试验,随机纳入了412例CRC患者,分别接受手术治疗或手术联合疫苗治疗,共随访7.6年,结果显示DFS和OS差异无统计学意义;进一步亚组分析发现Ⅱ期患者的OS和DFS确实有一定程度改善。随后,研究者为进一步探索OncoVax对Ⅲ期CRC患者的临床疗效,通过联合5-氟尿嘧啶和亚叶酸钙显示出了良好的安全性,在此基础上开展了一项多中心Ⅲ期临床研究,结果值得期待。
粒细胞-巨噬细胞集落刺激因子(granulocytemacrophage colony-stimulating factor,GM-CSF)基因转染的肿瘤疫苗(gene-transfected tumor vaccine,GVAX)是一种异基因全细胞疫苗,经过改造后可分泌GM-CSF。在一项针对pMMR晚期CRC患者的Ⅱ期临床研究中,GVAX显示出了抗肿瘤反应的调节作用。随后研究者对GVAX疫苗进一步改善,在临床前和临床研究中均尝试了表观遗传疗法以达到增强免疫活性的目的(NCT 01966289)。
随着癌症发病率和死亡率的不断增加,人们征服癌症的愿望也越来越迫切。新抗原疫苗在临床试验中对肿瘤治疗表现出明显的效果,有望成为未来缓解肿瘤发病率和死亡率不断上升的重要药物。它已经引起了免疫治疗领域专家的重视,是未来一个重要的发展方向。然而,新抗原的发展还存在一些制约因素,解决这些问题是新抗原能否得到广泛推广的关键。
通常在肿瘤样本中发现数千个非同义基因突变,但最终只有少数符合抗原标准。寻找更有效的抗原是一个需要解决的问题。研究表明,大多数特异性抗原倾向于分布在非编码区。近年来非编码研究的发展也将为新抗原的发现提供帮助。
缺乏有效的新抗原筛选方法是阻碍新抗原治疗发展的另一个障碍。目前,预测新抗原的算法正蓬勃发展。随着生物信息技术、人工智能和机器学习的发展,相信这个问题很快就会得到解决。
肿瘤在进化过程中是异质性的,因此可能发生突变的基因的每个部分都是不同的。因此,首先从患者身上获取局部肿瘤组织来预测该患者的新抗原可能是一个悖论。利用1~2种特异性新抗原对实体肿瘤组织进行完全识别和杀伤仍然是一个难题。
新抗原疫苗的开发周期长被认为是疫苗应用的主要障碍。开发周期长,导致研发成本增加,实验室和企业压力大,不利于疫苗临床应用。参与试验或治疗的患者生存期短,开发周期长,可能导致部分患者因药物开发周期长而无法接受最终治疗。目前,已经开发了许多方法来制备,配方和交付不同的癌症疫苗。然而,从技术上讲,这些疫苗需要在GMP条件下生产。最大的挑战,特别是对mRNA/DNA等小核酸疗法来说,是传递技术。基于新抗原的治疗大多是个性化的,从最初的基因测序到验证和生产,治疗成本可能非常高。成本仍然是最大的挑战。
(赵晓迪 聂勇战)
[1]MISSIAGLIA E,JACOBS B,D’ARIO G,et al. Distal and proximal colon cancers differ in terms of molecular,pathological,and clinical features[J]. Ann Oncol,2014,25(10):1995-2001.
[2]FAKIH M,VINCENT M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer[J]. Curr Oncol,2010,17 Suppl 1(Suppl 1):S18-30.
[3]FOLKMAN J. Tumor angiogenesis:therapeutic implications[J]. N Engl J Med,1971,285(21):1182-1186.
[4]MARGONIS G A,BUETTNER S,ANDREATOS N,et al.Association of BRAF mutations with survival and recurrence in surgically treated patients with metastatic colorectal liver cancer[J]. JAMA Surg,2018,153(7):e180996.
[5]PRAHALLAD A,SUN C,HUANG S,et al. Unresponsiveness of colon cancer to BRAF(V600E)inhibition through feedback activation of EGFR[J]. Nature,2012,483(7387):100-103.
[6]PYLAYEVA-GUPTA Y,GRABOCKA E,BAR-SAGI D.RAS oncogenes:weaving a tumorigenic web[J]. Nat Rev Cancer,2011,11(11):761-774.
[7]PIETRANTONIO F,DI NICOLANTONIO F,SCHROCK A B,et al. ALK,ROS1,and NTRK rearrangements in metastatic colorectal cancer[J]. J Natl Cancer Inst,2017,109(12):1-10.
[8]KEIR M E,BUTTE M J,FREEMAN G J,et al. PD-1 and its ligands in tolerance and immunity[J]. Annu Rev Immunol,2008,26:677-704.
[9]GORDON S R,MAUTE R L,DULKEN B W,et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity[J]. Nature,2017,545(7655):495-499.
[10]ANDRÉ T,SHIU K K,KIM T W,et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer[J].N Engl J Med,2020,383(23):2207-2218.
[11]LARSON R C,MAUS M V. Recent advances and discoveries in the mechanisms and functions of CAR T cells[J]. Nat Rev Cancer,2021,21(3):145-161.
[12]BENMEBAREK M R,KARCHES C H,CADILHA B L,et al.Killing Mechanisms of chimeric antigen receptor(CAR)T cells[J]. Int J Mol Sci,2019,20(6):1283.
[13]LICHTENSTEM C R,NGU R K,SHALAPOUR S,et al.Immunotherapy,inflammation and colorectal cancer[J].Cells,2020,9(3):618.
[14]SAHIN U,TURECI O. Personalized vaccines for cancer immunotherapy[J]. Science,2018,359(6382):1355-1360.
[15]WELLS D K,VAN BUUREN M M,DANG K K,et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction[J].Cell,2020,183(3):818-834.
[16]TRAN E,TURCOTTE S,GROS A,et al. Cancer immunotherapy based on mutation-specific CD4+T cells in a patient with epithelial cancer[J]. Science,2014,344(6184):641-645.
[17]SCHUMACHER T,BUNSE L,PUSCH S,et al. A vaccine targeting mutant IDH1 induces antitumour immunity[J].Nature,2014,512(7514):324-327.
[18]YANG W,LEE K W,SRIVASTAVA R M,et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses[J]. Nat Med,2019,25(5):767-775.
[19]KESKIN D B,ANANDAPPA A J,SUN J,et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial[J]. Nature,2019,565(7738):234-239.
[20]MIAO L,ZHANG Y,HUANG L. mRNA vaccine for cancer immunotherapy[J]. Mol Cancer,2021,20(1):41.