
下载掌阅APP,畅读海量书库
立即打开



结直肠癌(colorectal cancer,CRC)的发生是一个多步骤的过程,从正常上皮转变为腺瘤中间体,最终发展为腺瘤是多种遗传事件共同作用的结果。Fearon和Vogelstein提出CRC的发生是癌基因的激活突变和抑癌基因的失活突变共同作用的结果,且恶性肿瘤的形成至少需要4~5个突变。癌症基因组图谱(the cancer genome atlas,TCGA)对97例CRC患者进行的低丰度全基因组测序结果表明,16%的CRC样本是高突变的,并且其中3/4携带有高甲基化和 MLH1 沉默相关的MSI,1/4表现为体细胞错配修复基因和聚合酶ε( POLE )突变。常见的突变基因包括 APC 、 P53 、 SMAD4 、 PIK3CA 、 KRAS 、 SOX9 、 FAM123B 和ARID1A。
RAS是一类被称为GTP酶的蛋白,在人类大多数细胞中均有表达,其主要功能是细胞内信号转导,控制细胞增殖、细胞分化、细胞黏附、细胞凋亡和细胞迁移等过程。 RAS 突变时,促进细胞侵袭和转移。Vogelstein等首次发现CRC中存在 RAS 基因突变。 RAS 突变是CRC中的一个重要的体细胞突变, RAS 家族主要是 KRAS 突变。在发生转移的CRC样本中,约40%的CRC样本携带 KRAS 突变,主要发生在2号外显子的12(70%~80%)和13(15%~20%)位密码子。此外,3号外显子的59~61位密码子和4号外显子的117和146位密码子也可能发生 KRAS 突变。在2号外显子中,突变常见于密码子12:G12D、G12V和G12C,在3号外显子中突变的密码子为Q61H和Q61R,4号外显子中突变的密码子为A146T和A146V。 RAS 基因突变可能是结肠癌发生的启动事件。除了 RAS 这种点突变,癌基因还可通过扩增或重排被激活。研究发现在原发性CRC及其衍生细胞系中存在 neu 、 c - myc 或 c - myb 扩增。
BRAF 参与丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)通路激活,促进细胞生长、增殖和分化,在细胞迁移、凋亡和生存中也发挥重要作用。约90%的 BRAF 突变发生在第15号外显子T1799位点上(V600E)。这种颠换通过调节磷酸化,使 BRAF 活性比WT提高约10倍。 BRAF 突变的CRC患者是一群小而独特的群体,占所有CRC患者的8%~12%。约60%的 BRAF 突变肿瘤是低分化的,只有36%的 BRAF 突变肿瘤是高或中度分化的。黏液亚型肿瘤与 BRAF 突变的相关性更高(22%~67%)。不同于大多数CRC, BRAF 突变的CRC更常发生腹膜转移,较少发生肺和肝脏转移。 BRAF 突变和肿瘤中某些分子的相关性已被研究。 BRAF 和 KRAS 突变是互斥的,而13%的CRC患者中同时存在 BRAF 和 PIK3CA 突变,22%的CRC中发现 BRAF 和 PTEN 共突变。 BRAF V600E突变在高MSI患者(38.9%)中的发生率高于低MSI患者(9.3%)。 BRAF 突变与转移性CRC患者的预后负相关。
在CRC中,经常发生特定染色体区域的丢失。这种丢失通常发生在正常细胞的两条亲本染色体中的一条,该丢失区域通常含有抑制生长和分化的抑癌基因。CRC中的染色体丢失首次通过细胞遗传学方法被发现,后来研究者通过探针检测限制性片段长度多态性,确定两个亲本等位基因中是否有一个在肿瘤DNA中特异性丢失。CRC形成的遗传易感性发生在常染色体显性综合征和家族性腺瘤性息肉病。家族性腺瘤性息肉病的基因座已被定位到5q染色体。在无息肉病的患者中,有20%~50%的CRC和约30%的结直肠腺瘤存在5q染色体的等位基因丢失。不过,在家族性腺瘤性息肉病患者的腺瘤中,5q染色体的等位基因丢失是罕见的。超过75%的CRC中存在17p染色体的大片段丢失,但这种染色体缺失在各阶段的腺瘤中均相对少见。此外,研究发现在一些患者中,17p等位基因的丢失与腺瘤到癌的进展有关。CRC 17p染色体最常见的缺失区域包含 P53 基因。 P53 基因的一个等位基因点突变加上另一个野生型等位基因的丢失在CRC中频繁发生。此外,越来越多的研究发现,野生型 P53 可以抑制结肠癌的生长。CRC中第二常见的等位基因丢失位置是染色体18q,该丢失存在于超过70%的癌变和近50%的晚期腺瘤。在18q染色体上发现了共同的缺失区域,并且该区域存在抑癌基因——结肠癌缺失基因(deleted in colon cancer,DCC),该基因编码与细胞黏附家族分子高度同源的蛋白质。 DCC 基因在正常的结肠黏膜中表达,而在大部分CRC中低表达或不表达。这些表达缺失在某些情况下与 DCC 基因的体细胞突变有关。18q染色体上 DCC 基因位点的丢失,可导致遗传性非息肉病性CRC的易感性。 APC 基因在家族性腺瘤性息肉病和散发性CRC中的突变是一个关键的早期事件。除了上述提到的染色体5q、17p和18q等位基因丢失,在CRC中还存在1q、4p、6p、6q、8p、9q和22q染色体部分区域的丢失。
除了基因突变和染色体特定区域丢失,在CRC中还存在一些其他改变,如 ERBB2 和 IGF2 的基因组扩增, NAV2 和 TCF7L1 的融合,以及myc介导的转录激活或抑制。此外,肿瘤免疫微环境对CRC的发生也至关重要。高突变微卫星高度不稳定肿瘤表现出一种新抗原触发的细胞毒性免疫浸润,这有助于其对免疫治疗的反应性。然而,大量低突变负荷的结直肠癌似乎通过不明确的机制表现出激活的免疫微环境。
CRC中至少存在4种基因组或者表观遗传学不稳定性改变,分别是MSI、染色体不稳定(chromosomal instability,CIN)、CpG岛甲基化表型(CpG island methylator phenotype,CIMP)和广泛低甲基化。其中CIMP和广泛低甲基化详见本章第七节,本节重点介绍CIN和MSI。
基因组不稳定的最常见形式是CIN,超过85%的CRC中存在CIN。CIN是指染色体的数量异常或多重结构畸变。引起CIN的机制主要有如下三种:①由于监测染色体分离的信号通路缺陷所致。有丝分裂检测点是细胞周期的主要调控机制,它通过延迟分裂确保染色体分离的高保真度,直至所有染色单体对都正确地排列在中期板上。检测点信号的缺陷导致染色体错误分离和随后的非整倍体,异常数目的染色体分布到子细胞。②端粒功能障碍可驱动CIN,端粒是DNA-蛋白质复合物,由六聚体重复序列(人类的TTAGGG)组成,在染色体分离过程中保护真核生物染色体的末端不发生融合和断裂,当端粒末端保护受损时,染色体末端进入断裂-融合-桥循环,可以持续多代细胞,并导致基因组重组。③ DNA损伤反应。机体通过级联反应介导细胞周期阻滞来修复DNA损伤,在损伤无法修复的情况下,则诱导细胞衰老或凋亡。一些DNA修复蛋白参与人类癌症。一项体内研究证实DNA损伤反应和CIN直接相关。组蛋白H2AX是ATM和ATR的底物,单倍剂量不足可导致基因组不完整。
微卫星DNA是指基因组的一类短串联重复序列,几乎存在于各种生物的基因组中。MSI指由于插入或缺失突变引起的微卫星序列长度改变的现象。MSI存在于约15%的转移型结肠癌中。目前,许多临床实验室使用5种单核苷酸标记(BAT-25、BAT-26、NR-21、NR-24和MONO-27)评估MSI,这些检测方法均具有较高的灵敏度和特异度。携带MSI的结肠癌患者比携带CIN的结肠癌患者预后好,MSI和CIN通常是互斥的,几乎不会同时存在于同一种肿瘤。
MSI的潜在机制研究已经较为清楚,主要是由于DNA错配修复中的( MMR 家族)基因失活,可能是通过CIMP引发启动子甲基化失活,也可能是通过体细胞突变失活。此外,林奇综合征(遗传性非息肉病性CRC)患者几乎完全发展为MSI CRC,因为他们携带有 MMR 基因的生殖系突变,包括 MLH1 、 MSH2 、 MSH6 和 PSM2 。相反,散发性MSI CRC通常由于异常甲基化导致 MLH1 沉默进而导致 MMR 活性丧失。现在的研究发现,散发性MSI肿瘤与锯齿状瘤形成途径有关,通常携带 BRAF V600E 突变,而 MMR 基因(林奇综合征)生殖系突变导致的癌症没有 BRAF 突变。
CRC分子演进过程中的最经典模型是正常上皮-腺瘤-腺癌的多阶段基因假说模型,该模型于1990年由Fearon和Vogelstein提出,约85%的CRC由该演进过程形成。首先, APC 抑癌基因失活导致异常隐窝灶形成,随后染色体18q杂合性缺失, KRAS 、 P53 基因突变等CIN事件和基因突变的累积驱动腺瘤形成、进展、癌变(图3-1-1)。
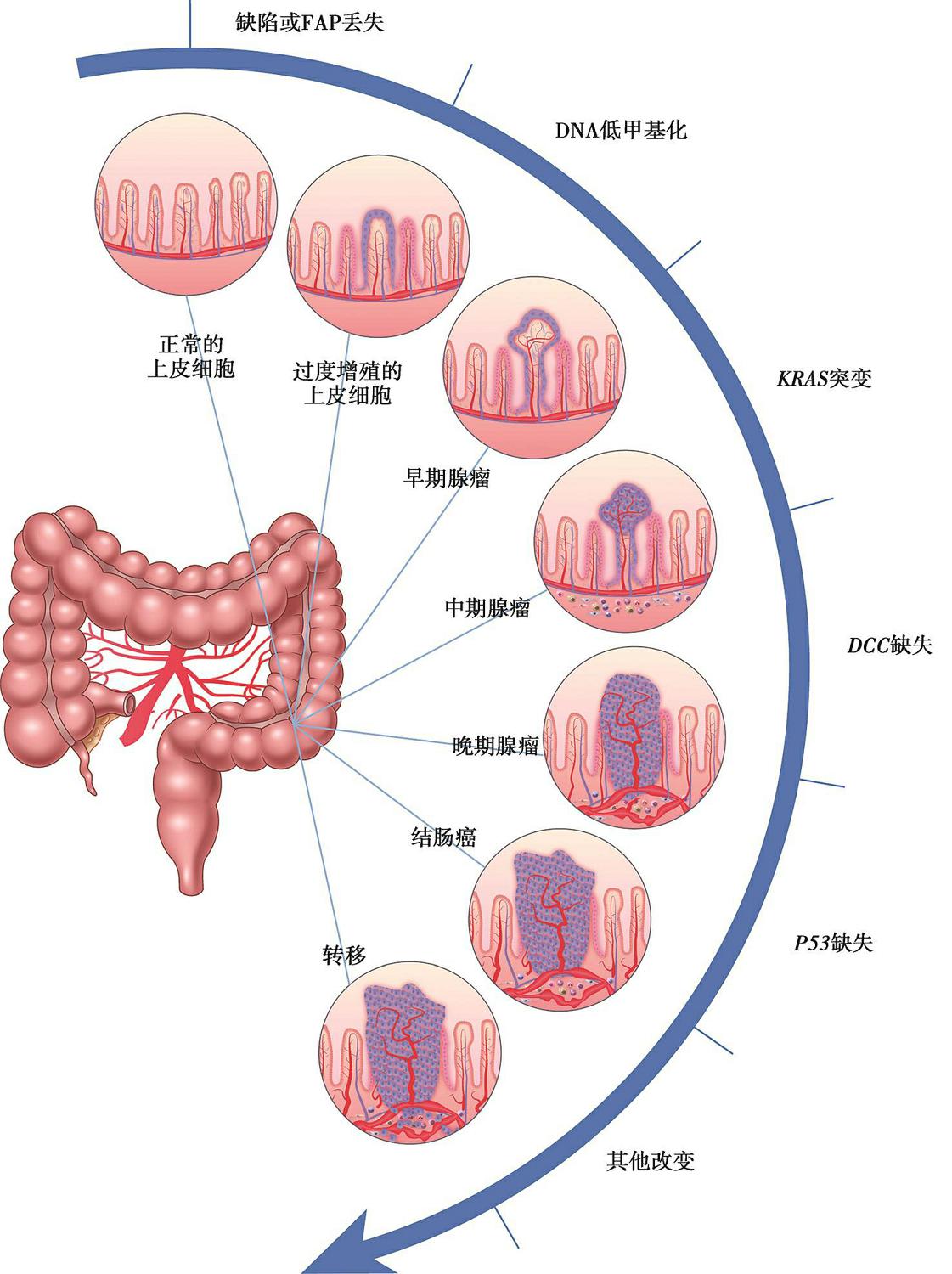
图3-1-1 结直肠癌发生的遗传模型
肿瘤发生需经过一系列的基因改变,其中包括癌基因( RAS )和抑癌基因(特别是5q、17p和18q染色体上的基因)。5q突变或FAP丢失是CRC发生的早期事件,随之是甲基化改变, KRAS 突变紧随其后,接着是18q丢失,最后是17p丢失。
Chen B等通过单细胞RNA-Seq、多重免疫荧光和多重免疫组织化学绘制了两种人类最常见的CRC息肉-常规腺瘤和锯齿状息肉的单细胞转录组图谱和成像图谱。他们发现传统腺瘤中干细胞标志物OLFM4和SOX9高表达,且 LGR5 、 OLFM4 、 ASCL2 、 AXIN2 、 RNF43 和 EPHB2 等Wnt通路相关基因被激活,是由Wnt通路驱动干细胞扩张而产生。而锯齿状息肉高表达MUC5AC,其形成的原因可能是肠腔表面反复刺激导致细胞化生进一步形成,而非隐窝起源。
除了经典的“正常上皮-腺瘤-腺癌的多阶段基因假说模型”,通过基因组学分析,对比同一患者身上的原发性肿瘤和转移灶,又提出5种肿瘤向转移发展模式模型(图3-1-2):线性进化、“大爆炸”模型、合作进化、克隆选择、转移特异性突变。

图3-1-2 肿瘤演进模型
线性进化是指肿瘤由最初的分子特征所决定的路线进行演化。这些分子特征包括驱动突变、转录活性情况、组成原发性肿瘤的细胞类型等。这意味着肿瘤最初的突变决定了它是否会向着转移发展,而后续发生的突变影响都是十分有限的。这样,肿瘤的发展情况从一开始就是可以预测的。
“大爆炸”模型是指正常组织在转化为肿瘤后,会经历一个高度的基因组不稳定阶段,产生具有各种分子变异的克隆细胞群体,形成高度的肿瘤异质性。在之后肿瘤的发展过程中,部分亚克隆群体稳定下来并且相对平稳发展,从而形成高度异质的原发癌,并形成转移。在这一模型中,肿瘤的转移仍是可预测的。
合作进化认为肿瘤转移的形成并不是某一突变类型的肿瘤细胞就能够形成的,而是需要有多种具有不同分子变异的原发癌亚群共同实现转移性的获得。对于合作进化,特定的突变类型组合形成转移仍是可以确定的,因此肿瘤是否会形成转移仍是可以预测的。
克隆选择则更多地关注单细胞的变异,认为肿瘤中各种克隆群体中一个或几个具有额外突变的单细胞克隆被正向选择,并且相对于肿瘤主体细胞克隆群体的分子组成,这些获得额外突变的单细胞克隆独立地向转移发展并最终形成转移。基于该模式,单细胞层次的分析仍能预测肿瘤转移形成的可能性。
转移特异性突变认为肿瘤初始的传播是由原发性肿瘤的变异所决定的,这些突变的肿瘤细胞在初始传播后又获得额外的突变,从而导致肿瘤的微转移形成临床上明显可见的转移性肿瘤。即转移决定性的变异是在脱离原发性肿瘤后获得的,基于这一点,肿瘤转移性是无法通过分析原发性肿瘤的基因情况来获得的。
CRC从非典型增生发展为原发癌,侵袭周围组织,最终转移到淋巴结和远端组织。这些模型对通过分析原发肿瘤预测远处转移的可能性做出不同的说明。①线性进化:初始驱动突变决定转移风险,额外突变对转移风险影响有限;②大爆炸模型:在转化后,产生具有各种分子病变的克隆,一些克隆向转移方向线性进化;③协同进化:具有不同分子病变的原发癌需要不同亚群来完成转移;④克隆选择:经典的肿瘤进化模型预测原位癌中获得额外突变的亚克隆的选择,连续的癌变步骤需要额外的突变来提供选择性优势;⑤转移特异性突变:最初的播散是由原发癌中存在的突变决定的,在播散后获得额外的突变,使微转移成长为临床上明显的转移肿块。
这些模型有可能同时存在。原发性肿瘤的驱动突变可能会决定肿瘤是否向转移发展,但是原发性肿瘤中不存在或只存在于某一亚群中的额外基因也可能会对转移的发生产生影响,只是这些额外基因的变化可能会因驱动突变的不同而变化。另外也有报道发现具有不同突变的细胞在至少一个转移形成步骤中合作的模式,这与合作进化模型一致。
值得一提的是,对人类癌症的深度测序支持“大爆炸”模型。“大爆炸”模型关注肿瘤细胞演化时分支突变是否改变该分支继续向转移方向演化的潜力。基于甲基化模式的腺瘤隐窝系谱分析指出多数克隆出现在腺瘤发展的早期,形成高度的瘤内异质性,而克隆的稳定性随着时间提升。CRC基因组分析也指出多数变异,都是在致癌早期形成的,并且这些变异会在肿瘤形成的过程中缓慢均匀地分布到整个肿瘤中。这些都符合“大爆炸”模型的肿瘤演化模式。
这样的“大爆炸”模型意味着在肿瘤发展的早期大量异质亚克隆群体产生,然后与复制出错相关的突变连续积累。肿瘤发展后期出现的变化只存在于肿瘤的小区域。肿瘤转移的潜能也在肿瘤发展较早的时期就已经决定。在早期产生的一些克隆克服了基因不稳定,并成长为晚期肿瘤以及形成转移灶。因此,大爆炸模型通过突出分支事件的时间结合了分支模型。基于这样的模式,转移灶和原发灶会有许多一致的突变,而某一个细胞亚群中某些突变的缺失意味着这个亚群可能是转移灶的祖细胞。
也有研究指出并非所有细胞都存在原发性肿瘤带有的突变。在他们的研究中观察到在转移细胞中 KRAS 的流行率最高,达到56.9%。这样的观察结果比较难以融入线性进化模型中。这一研究成果意味着由多种细胞组成的转移瘤可能来自不携带突变基因的原发肿瘤而在转移处获得新的突变。这一研究成果可能需要通过转移特异性突变模型或合作转移模型予以解释。
CRC显示出明显的形态学演变,从腺瘤上的异常隐窝病灶到浸润性癌和转移,但这种演变的分子相关性不是线性的,而是呈现分支性的。CRC的形成主要由于基因组不稳定,原癌基因的驱动突变可能对肿瘤的发展和转移起重要作用,肿瘤发展过程中额外的基因突变对肿瘤演化的贡献尚不明确,但是其影响很可能有限。
在众多肿瘤演化模型中,“大爆炸”模型较好地解释了CRC的突变模式。在肿瘤形成的早期获得众多突变克隆群体,表现高度的瘤内异质性。随后部分克隆群体稳定下来,并经历一个相对平稳的生长阶段。原发性肿瘤已经拥有肿瘤进化的大部分决定因素,这与通过分析原发性肿瘤来预测结果的可能性一致。后来发生的乘客突变、个体突变只是有限的拓展。
(周天华 刘祥瑞)
目前认为,CRC在其发展的不同阶段,可以出现多种基因异常,除少数遗传性肿瘤外,在CRC发生发展过程中,需要众多基因改变的相互作用,如 APC 、 c - myc 、 P53 、 P16 、 DCC 、 MCC 、 DPC4 、 BRAF 或错配修复基因等。
肠上皮细胞不断更新,并受到多种信号通路的严格调控。这些通路的突变可导致上皮细胞生长受阻、凋亡延迟或失败,促进肿瘤的形成、生存、血管生成和转移。为使其作为治疗靶点来对抗CRC,对这些通路的理解仍需不断深入,了解这些机制也有助于预防肿瘤的形成。
CRC中与Wnt信号通路相关的主要基因突变为腺瘤性结肠息肉 APC 突变。 APC 为抑癌基因,其编码的蛋白负调控Wnt信号通路,调控细胞增殖、分化。Wnt/β联蛋白通路是高度保守的,在干细胞分化和细胞生长中起着关键作用,对胚胎发生至关重要。Wnt蛋白是生长刺激因子,当APC功能异常时,可通过上调β联蛋白,激活促进细胞增殖的基因如 MYC 、 cyclin D1 的转录,使细胞异常增殖,形成肿瘤。因此,该通路的改变参与驱动了肿瘤的发生发展。CRC中Wnt信号通路的改变也能影响紧密连接,这导致细胞黏附性降低,从而有利于肿瘤细胞的迁移和转移。此外,Wnt信号通路的异常并非唯一,含有 APC 突变的肿瘤也可能出现其他通路的改变。 APC 是最常见的突变基因,且由于CRC中 APC 突变频率高且广泛,因此 APC 突变并不能很好地反映患者的预后,即与患者的预后无明显相关。
哺乳动物共有19个 Wnt 基因,它们都在细胞命运决定、细胞周期、增殖和迁移等多个发育过程中发挥调控作用。细胞膜表面细胞受体包括细胞表面卷曲(Fz)和低密度脂蛋白受体相关蛋白(LRP)复合物。与此同时,还存在一个由多种蛋白组成的细胞内复合物,包含β联蛋白、dishevelled(Dsh)、Axin和糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3)等。该蛋白复合物通过蛋白酶体降解β联蛋白,从而调节细胞内β联蛋白的水平。在β联蛋白磷酸化和泛素化后,β联蛋白被细胞蛋白酶体降解。当蛋白复合物与配体结合时,β联蛋白的降解过程被抑制,导致细胞中活性磷酸化的β联蛋白积累。随后,β联蛋白进入细胞核,并诱导激活促进细胞增殖的基因如 MYC 、 cyclin D1 转录。因此,Wnt过表达可以促进肿瘤生长,Wnt/β联蛋白通路的异常可以导致CRC的发生。具核梭形杆菌通过诱导Wnt/β联蛋白调节分子 Annexin A1 促进CRC。该通路的发现,提供了靶向Wnt/β联蛋白通路的小分子药物的潜在思路。
受体酪氨酸激酶(receptor tyrosine kinase,RTK)是胞外信号传递到胞内的重要途径之一,RTK在多种细胞行为中发挥重要作用,如细胞增殖、分化、细胞代谢、迁移、细胞周期控制等,RTK的突变与肿瘤密切相关。
表皮生长因子受体(epidermal growth factor receptor,EGFR)存在于细胞表面,具有细胞外配体结合域。表皮生长因子(epidermal growth factor,EGF)作为一个配体与EGFR结合,导致RTK二聚化,从而激活RTK活性,二聚化的RTK发生自磷酸化,并激活下游诸如RASMAPK信号通路等多条信号转导途径。Seven-less蛋白(Son of sevenless,Sos)是一种鸟嘌呤核苷酸交换因子,可以使RAS-GDP转换为有活性的RAS-GTP。一个接头分子Grb-2通过其SH2结构域与被磷酸化的酪氨酸残基相互作用,然后通过Grb-2的SH3结构域与Sos相互作用。活化的RAS蛋白启动RAS-丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)磷酸化级联反应,通过磷酸化依次激活促分裂原活化的蛋白激酶激酶激酶(mitogen-activated protein kinase kinase kinase,MAPKKK)、促分裂原活化的蛋白激酶激酶(mitogenactivated protein kinase kinase,MAPKK)和MAPK或胞外信号调节激酶(extracellular signal-regulated kinase,ERK)。ERK通过靶向细胞质底物或核底物调控细胞活动,如细胞的增殖和存活。细胞质底物包括c-fos和c-Jun(由MAPK二聚形成),它们进入细胞核,与DNA的AP-1基序相互作用,启动转录。ERK也可磷酸化核糖体S6激酶。S6蛋白可以完成两种功能,一是负调控Sos分子(通过抑制GDP向GTP转化而关闭信号通路),二是进入细胞核调控CREB转录因子。MAPK也可能直接调控核底物MYC。该途径的失活可通过GAP蛋白水解GTP发生。
RAS-MAPK通路参与各种细胞过程,如细胞的生长、增殖和存活。该通路的异常调控可刺激肿瘤细胞的生长、存活、血管生成和转移。转移性CRC几乎占新诊断CRC病例的一半,预后较差,而EGFR是转移性肿瘤细胞增殖、分化和生存的关键因素。KRAS是RAS信号蛋白家族的一员,在EGFR介导的细胞增殖和生存调节中发挥着重要作用。在约40%的CRC病例中, KRAS 基因的突变在癌症早期即存在。研究发现,与携带 KRAS 突变的患者相比,野生型 KRAS 患者的总生存期、无进展生存期和/或缓解率均显著更高。据报道,在CRC病例中,EGFR异常调控、扩增、拷贝数增加和过表达促进MAPK激活,正作为一个可能的、有希望的治疗靶点进行研究。
磷脂酰肌醇3-激酶(phosphoinositide 3-kinase,PI3K)是细胞内脂质激酶成员之一,参与调控细胞增殖、分化和存活。PI3K-AKT-mTOR信号通路的过表达在各种形式的癌症中都有报道,尤其是CRC。由于它们在CRC的起始和进展事件中发挥着重要作用,因此被认为是一个潜在的治疗靶点。RTK与配体结合后,会自动磷酸化并激活PI3K,PI3K磷酸化脂质蛋白,使磷脂酰肌醇4,5-双磷酸变为磷脂酰肌醇3,4,5-三磷酸(phosphatidylinositol 3,4,5-triphosphate,PIP3)。PIP3信号蛋白,如3-磷酸肌醇依赖性蛋白激酶1(3′-phosphoinositide-dependent kinase 1,PDK1),通过作用于丝氨酸和苏氨酸残基激活蛋白激酶B(AKT/PKB)。AKT靶向下游蛋白,如哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),负责细胞周期进展、增殖、延迟凋亡、生长和存活。PTEN通过去磷酸化PIP3下调通路。
PI3K-AKT通路的异常表达在CRC的生长和发展中起重要作用。该通路的异常表达(无法关闭)导致细胞持续和不受抑制地生长和存活,例如 PTEN 突变,突变的 PTEN 能促进不受控制的细胞生长,从而诱导肿瘤的发生。据统计,PI3K异常表达占人类癌症的30%。磷酸化AKT过表达与70% CRC患者细胞分裂和凋亡抑制以及 PTEN 表达异常有关。AKT靶向的下游蛋白mTOR还被证明有利于血管生成和生长,使用阿司匹林(mTOR抑制剂)能够抑制CRC的进展。下调基因 PHLDA2 可抑制肿瘤生长,促进细胞自噬,抑制上皮-间充质转换(epithelial-mesenchymal transition,EMT),其机制可能与PI3K-AKT-mTOR和PI3K-AKT-GSK-3β信号通路有关。
血管生成对癌症的发生、细胞增殖、生长、侵袭和转移起着至关重要的作用。各种促血管生成和抗血管生成因子调节血管生成,如血管内皮生长因子(vascular endothelial growth factor,VEGF)、成纤维细胞生长因子(fibroblast growth factor,FGF)、转化生长因子-α(transforming growth factor-α,TGF-α)、TGF-β、血小板生长因子(plateletderived growth factor,PDGF)和血管生成素,这些因子被释放到肿瘤微环境中参与肿瘤血管形成。VEGF家族及其受体是血管生成最重要的调节因子。VEGF蛋白家族由5个蛋白组成——VEGF-A、VEGF-B、VEGF-C、VEGF-D和胎盘生长因子(placental growth factor,PIGF)。这些蛋白质与内皮细胞上的3种受体酪氨酸激酶受体——血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)1、2、3及2种非酪氨酸激酶辅受体——神经营养因子受体1、2结合。VEGF与VEGFR、VEGF-A、VEGF-B、PIGF之间的多样化网络最终能够促进血管生成。其中,VEGF-C和VEGF-D主要参与淋巴血管生成。VEGF-A和VEGF-B通过VEGFR 1和VEGFR 2显著地结合在内皮细胞和一些非内皮细胞上。VEGFR 3在内皮淋巴细胞上表达,并与VEGF-C、VEGF-D具有高亲和力。
VEGFR 1属于受体酪氨酸激酶家族蛋白,目前已知表达于内皮细胞、炎症细胞和肿瘤细胞。在早期血管生成事件中,VEGFR 1主要调控内皮细胞的分化和细胞迁移,促进上皮细胞的分化,而对细胞增殖的作用并不显著。此外,VEGFR 1的激活介导了几个下游通路的激活,如炎症细胞中的PI3K-AKT-MAPK/ERK,导致炎症细胞因子和白介素介导的上调,如TNFα、IL-1β、IL-6和IL-8,进而诱导细胞迁移。VEGFR 1的功能尚不完全清楚,主要在血管生成过程中起调节作用。
VEGFR 2主要表达于血管和淋巴管内皮细胞,是一个200~230kDa的蛋白,参与血管形成。VEGF-A结合到VEGFR 2并使之激活,导致多个下游通路的激活,如RAS-RAF-ERK/MAPK和PLCγ,进而促进细胞生长。VEGFR 2的激活也能激活PI3K-AKT信号通路,从而调控细胞死亡。
VEGF-C、VEGF-D与VEGFR 3的结合能导致淋巴管形成。激活的VEGFR 3激活下游RAS-MAPK-ERK和PI3K-AKT/PKB通路,导致淋巴内皮细胞的分化、增殖、存活和迁移。
有足够的证据表明,VEGF水平和VEGFR活性升高,与CRC预后不良相关。VEGF水平升高在早期和晚期的CRC患者中均有发生。在CRC中, KRAS 、 P53 、 COX - 2 突变和缺氧均能导致细胞生长和迁移,从而调控VEGF-VEGFR信号通路。在肿瘤原发部位,VEGF/VEGFR复合物的促血管生成功能促进了肿瘤的发展和转移;在转移部位,则能促进新血管形成,进而在促进癌症生长和存活中都起到重要作用。抑制细胞内VEGF信号通路,通过调控参与细胞运动的蛋白,也能够显著抑制CRC肿瘤细胞的迁移和侵袭。因此,抗VEGF或抗VEGFR治疗,可减少肿瘤形成和转移。
So-Yeon Park等通过分析包含CRC患者数据的公共数据库,发现JAK-STAT信号在放疗耐受的CRC组织中被激活,且与肿瘤的局部侵袭和远处转移相关。JAK2在CRC干细胞亚群中被优先过表达,并伴有STAT蛋白的磷酸化,尤其是信号转导及转录激活蛋白3(signal transducer and activator of transcription 3,STAT3)。JAK2-STAT3信号通路通过限制细胞凋亡和增强克隆潜能,在促进肿瘤起始和放射抵抗中发挥重要作用。在机制上,STAT3直接结合周期蛋白D 2 启动子增强了CCND2的转录。CCND2的表达对通过维持完整的细胞周期和低水平的DNA损伤积累的持续性癌症干细胞生长是必需的。
四种Janus激酶(Janus kinases,JAK)包括JAK1~3和TYK2,与结肠中存在的细胞因子受体存在相互作用。虽然细胞因子受体有多种,但JAK在细胞内信号通路中有共同的机制,包括信号转导和激活STAT蛋白,从而调控下游基因的表达。当细胞因子与受体结合后,受体构象发生改变,形成二聚体,受体二聚化使JAK相互交叉磷酸化,成为激活的JAK。活化的JAK进一步磷酸化细胞因子受体的胞内段酪氨酸残基。磷酸化的酪氨酸残基被具有SH2结构域的STAT蛋白识别并结合,进而STAT的C端酪氨酸被结合在受体上的JAK磷酸化。磷酸化的STAT从受体上解离下来,2个磷酸化STAT蛋白的SH2结构域相互结合,形成二聚体,并暴露核定位序列,进而STAT蛋白入核,识别并结合γ活化序列元件,引起促炎基因表达和转录,并在炎性肠病的发病机制中发挥作用。抑制JAK功能的药物结合并阻止其磷酸化,有效地阻断该途径。JAK蛋白还可以向PI3K蛋白(AKT通路)和RAS蛋白(MAPK通路)发起信号。
环状RNA circSPARC通过调控JAK-STAT通路增强CRC的迁移和增殖。长链非编码RNA(lncRNA)RP11-468E2.5能够靶向 STAT5 和 STAT6 ,通过JAK-STAT信号通路抑制CRC细胞增殖,刺激细胞凋亡。CPEB3(cytoplasmic polyadenylation element binding proteins)作为RNA结合蛋白结合 JAK1 mRNA的3′UTR抑制JAKSTAT通路,在CRC细胞中发挥作用。CPEB3通过转录后调控JAK-STAT信号通路在CRC中发挥抑癌作用。敲低CPEB3可激活JAK-STAT信号通路,从而触发CRC细胞的增殖和转移能力。趋化因子CXCL1可受miRNA miR-302E调控,使JAK-STAT信号通路失活,进而影响CRC肿瘤细胞的增殖、迁移、侵袭和凋亡。
CRC的发展是一个多步骤的过程,包括一系列随时间积累的遗传、组织学和形态学改变。此外,根据突变来源的不同,CRC可分为家族性、散发性和遗传性CRC。这些突变还可能导致染色体畸变、MSI、CIMP和CIN。这些突变会导致影响癌症进展的各种途径的信号通路异常,它们包括PI3K-AKT、WNT、TP53和MAPK通路等,还包括HGF/cMET信号通路、TGF-β信号通路、Notch信号通路、SHH信号通路、Hippo信号通路、KEAP1/Nrf2信号通路等(图3-2-1),信号通路的任何异常,无论是通过功能缺失性突变还是功能获得性突变,都可能导致结肠癌的发展,增大肿瘤的发生频率。
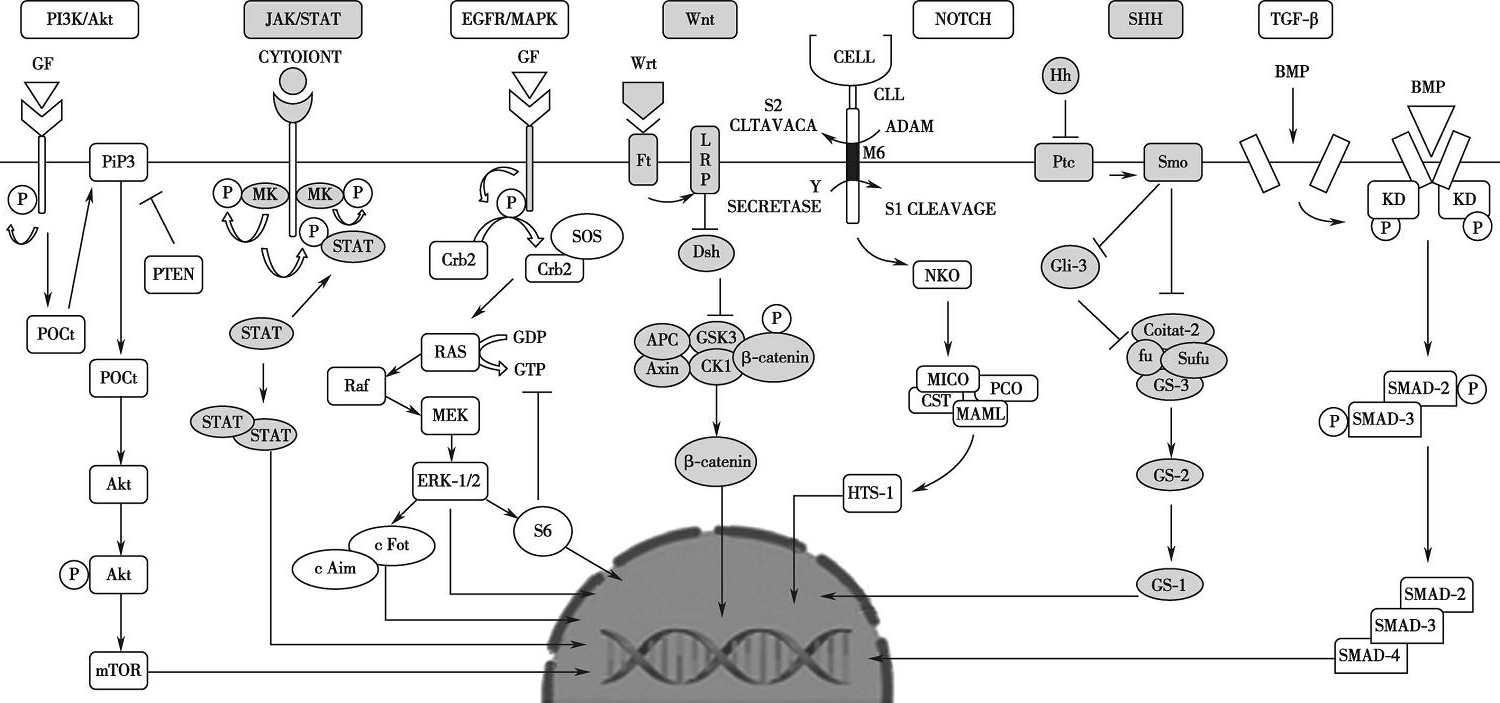
图3-2-1 CRC中相关的部分信号通路
CRC因其在世界范围内的高发病率和高死亡率,已成为全球性的公共卫生问题。CRC是全世界第三大常见恶性肿瘤,常见于欧洲、北美以及新西兰等地区。从世界范围看,中国是结直肠癌的低发区,但目前在中国结直肠癌已是名列发病率第三位的常见恶性肿瘤。其发病率呈上升趋势,尤其是结肠癌发病率增长速度迅猛。
CRC的发生和发展是由遗传和环境因素共同诱发的,对其研究及建立新的预防策略至关重要,这是阻止其发病率增加的最重要的措施之一。在过去的10年中,已经确定了主要的结肠癌基因,它们的突变与CRC的高易感性相关。此外,对它们遗传模式的研究发现了遗传性CRC综合征,现已可以进行基因诊断,开展针对性的风险管理项目和遗传咨询,可以应用于患者及其亲属,减少罹患CRC的风险。虽然对于与CRC相关的大多数主要癌症基因已有了较为深入的认识,但这种疾病与其他环境因素的关系仍不明确。
在此背景下,仍需要对饮食、微生物群与CRC的关系进行更深入的研究。这些研究的结果可能有助于通过改变生活方式来降低罹患CRC的风险。此外,由于肠道微生物群的组成可能也影响CRC的发展,基于益生菌和益生元的辅助治疗仍处在研究中,以期提高对传统化疗药物的反应,减少给药剂量和给药频率,从而改善患者的生活质量。同样,在可预见的将来,开发和实施新的特异性和更敏感的生物标志物将改善诊断策略,从而使临床医师能够在疾病的早期阶段发现CRC,从而改善患者的预后。
目前,只有肿瘤样本中微卫星DNA区域的不稳定性(instability of microsatellite DNA regions)和 KRAS 突变的测定被用于临床。对于CRC的早期诊断,目前正在评估基于miRNA表达、基因微阵列和CIMP的不同检测方法,尽管它们都有着很好的前景,但仍需要对更广泛的人群进行进一步的研究来验证。在精准治疗的浪潮下,个体化治疗手段正迅速成为临床医师不可或缺的工具。因此,有必要对每个患者的肿瘤特征进行深入分析,以找到最合适的治疗方法。
当前大部分的CRC研究集中在开发新的治疗方法上。这些新疗法与传统疗法相比更温和,更有效,也能更好地改善未来CRC患者的总体生存率和生活质量。
(郑小凤)
生命体需要将遗传物质可靠地传承到下一代,正常体细胞分裂也需要准确地将基因组进行复制,并且均分到两个子代细胞中,以确保子代细胞具有与亲代细胞完全一致的遗传物质。这种将基因组信息由亲代向子代,或由亲代细胞向子代细胞的可靠传递体现了基因组稳定性的特征,其含义包括遗传物质(DNA或RNA)的无差错复制、对复制错误或偶发DNA、RNA损伤的修复;以及分裂期遗传物质的正确分离。与之相反,如果复制过程发生了异常高频的突变,或是分裂或修复过程失败,则会造成子代细胞中各种形式的基因组改变。包括但不限于:基因突变、插入、删除,染色体片段重组,染色体易位,整条染色体重复或丢失,多倍体现象。因为各种原因造成的基因组信息易改变的倾向,即为基因组不稳定性。随着这些改变的积累,细胞的生命活动逐渐受到影响,可造成分裂障碍或生长失衡,甚至发生癌变。
基因组稳定性的维持主要依靠细胞分裂过程中4个方面的共同作用:① S期DNA复制的高保真性;②有丝分裂期染色体的准确分离;③各种DNA损伤和修复机制的有效运行;④细胞周期进行和细胞命运的正常调控。肿瘤发生的分子生物学进程可被视为多次细胞分裂过程中基因组改变的逐步积累(图3-3-1):具有增殖功能的祖细胞中,关键基因的改变将正常细胞转化为癌前细胞;某些癌前细胞中继发的基因组改变使它们获得继续分裂的优势,这些无限增殖的恶性细胞已经具备了癌细胞的特征;而分裂过程中调控机制的缺陷容许了基因组改变的进一步积累,导致具有更高侵袭性的癌细胞亚群的出现。因此,基因组不稳定性不仅是癌症的重要特征,而且是肿瘤发生的驱动力。

图3-3-1 基因组不稳定性在癌症发展中疾病干预中的作用
CRC的发生发展与基因组不稳定性密切相关。越来越多的证据表明,肿瘤细胞的遗传学特征不仅决定了肿瘤的发生、进展,也与预后及靶向治疗密切相关。统计结果表明,CRC病例中,60%~80%为不具有家族史或明显遗传因素的散发病例,剩下的20%~35%则是具有家族性或潜在的基因易感性的遗传性病例。与CRC密切相关的基因组不稳定性的形式主要有MSI及CIN。下文分别阐述其相关调控机制及发生原因。
微卫星序列是由短串联重复片段组成的重复性DNA序列,重复片段通常只有1~5个碱基对长度。MSI性是一种以微卫星序列中重复单元数量增多或减少为特征的基因组不稳定性形式。Bethesda指南推荐通过5个标志性的微卫星位点评估肿瘤基因组中的微卫星状态,包括2个单核苷酸位点(BAT26和BAT25),以及3个二核苷酸重复(D2S123、D5S345和D17S250)。30%及以上标记性位点展现出不稳定特征的肿瘤为MSI-H,小于30%标记性位点不稳定的肿瘤归类为MSI-L,而未出现明显不稳定性的肿瘤则归类为微卫星稳定肿瘤。约15%的人类CRC病例中可以观察到MSI,而3%~5%的CRC病例最终诊断为遗传性非息肉病性CRC。
MSI主要由DNA错配修复(mismatch repair,MMR)机制障碍而导致。错配修复机制是一种高度进化保守的修复系统,主要用于修复复制错误或者DNA损伤介质造成的碱基对错配。MMR在DNA复制和B细胞的种类转化重组(class switch recombination,CSR)过程中具有重要作用。DNA复制过程中,逃避了DNA聚合酶校对功能的碱基对错配,需由MMR系统来进行纠正。人类细胞中,MMR系统通过DNA链缺口识别新合成的DNA链,MSH2与MSH6或者MSH3分别形成MutSα或MutSβ复合体,首先识别并分别结合于错配碱基对或微卫星复制数错误形成的环状结构,MLH1与PMS2或PMS1构成的MutL复合体进一步结合于MutS复合体,激活其内切酶活性在DNA链上形成缺口。Exo1蛋白通过缺口进入,将错配部位的新生链切除。DNA聚合酶pol δ以切除后形成的单链为模板重新合成错配部位的DNA,之后经DNA单链断裂修复通路的连接酶Ligase1连接。MMR障碍可由MMR关键基因突变或表观遗传学机制导致。HNPCC与MMR基因 MSH2 、 MSH6 、 MLH1 、 PMS1 和 PMS2 等突变相关;而散发病例中的MMR系统失活,则多由于 MLH1 基因5’端启动子附近的CpG岛的异常超甲基化,导致其转录和表达障碍。
当MMR系统障碍时,DNA复制时造成的碱基对错配和MSI无法得到有效修复,遗留下大量MSI。研究表明某些特定的病理性肿瘤抑制因子突变与MSI息息相关。稀有情况下,某些微卫星序列位于关键性生长调节因子的外显子区域,MMR缺陷引起MSI区域序列长度变化,造成编码区框移,产生无功能的蛋白造成基因失活。已知发现的MSI相关抑癌基因突变包括 TGFBR2 、 IGF2R 、 BAX 、 ACVR2 、 SEC63 和 AIM2 等。这些基因的失活,造成细胞生长增殖的失控,开启了细胞癌变的潘多拉魔盒。
CIN是高频的染色体水平变化的基因组改变,包括染色体或大范围染色体片段的重复、丢失、重组、易位等,也包括多倍体现象。CIN可导致细胞与细胞间不同的可见核型改变。单个肿瘤中,某些CIN类型可以在所有肿瘤细胞中见到,而某些则具有细胞特异性,说明了CIN从细胞癌前病变开始,不断产生逐渐积累,贯穿于整个癌症的发生与发展过程中。体外的肿瘤细胞培养也可以观察到CIN的出现和逐渐积累,证明了CIN是结直肠肿瘤的特征性改变。约85%的CRC病例中可以观察到CIN。CRC中常见的CIN类型包括染色体臂1p、5q、8p、17p、18p、18q、20p和22q缺失,7号染色体和染色体臂1q、8q、12q、13q和20q重复,以及各种复杂形式的染色体易位等。
与MMR系统相对于MSI的高度特异性不同,CIN相关调控基因的鉴定极具挑战性。时至今日已有100种以上的基因证明具有CIN相关性。MUTYH-相关性息肉病(MAP)是与CRC高度相关的癌前病变,其病变细胞以具有高频的G:C-A:T突变为特征。该疾病患者 MYH (又名 MUTYH )基因具有双位点胚系突变,多为165位酪氨酸突变为半胱氨酸,或是382位点的甘氨酸突变为天冬氨酸。 MYH 基因是DNA碱基切除修复(base excision repair,BER)通路的糖苷酶(glycosylase)。DNA氧化损伤发生时,主要会在鸟嘌呤位点形成8-氧-鸟嘌呤(8-oxoguanine,8-oxoG),8-oxoG具有高度致突变性,在DNA复制时可被聚合酶错认为胸腺嘧啶与腺嘌呤配对。MYH可切除氧化造成的8-oxoG位点错误的腺嘌呤配对,故失活时造成大量的G:C-A:T突变;有丝分裂纺锤体检查点(mitotic spindle checkpoint)相关基因调控有丝分裂期纺锤体与动粒的相互作用,确保姐妹染色单体的正确分离。部分含有CIN的CRC细胞中鉴别出来含有 MAD 基因和 BUB 等基因突变。 MAD 和 BUB 基因产物控制检查点的激活和信号转导,突变时可造成姐妹染色单体分离障碍;中心体数目和功能相关的调控基因与CRC中多倍体的发生也密切相关,多项研究证实了CRC细胞中中心体相关丝氨酸、苏氨酸调节激酶STK15的高表达,MPS1和寿命蛋白(mortalin)等中心体数目调节蛋白,也被证实与CRC的多倍体基因组不稳定相关;细胞周期因子与细胞周期检查点相关蛋白CDC4、端粒酶相关蛋白TERT和RNA转录基因 TERC 等都会导致CRC中的CIN;除此之外,研究表明DNA双链断裂的同源重组(homologous recombination,HR)修复通路基因 ATM 、 BRIP1 、 POLD 、 RAD51 等的突变与区域进展期CRC的CIN高度相关。
与MSI相比,CIN带来的基因表达变化往往更多更复杂。除了区域性的突变、框移失活等,含有CIN的结直肠肿瘤往往还具有大量的染色体易位。染色体易位是在细胞失控增殖分裂过程中,DNA因各种原因由基因组中的多个脆弱位点发生断裂后,借由功能异常的DNA的双链断裂HR或者非同源末端连接(non-homologous end-joining,NHEJ)修复通路修复,而导致的异源DNA末端之间的相互连接。这种易位可以导致某种原癌基因被连接至上游的强启动子,从而大量表达,加速细胞生恶性化。而快速的分裂带来更多的CIN,形成恶性循环,进一步增加了肿瘤的侵袭力。
Fearon和Vogelstein在1990年提出了CRC进展过程中不同阶段的基因组改变,又称“Vogelgram”图(图3-3-2)。经后续研究结果补充后,修订后的图清楚展示了伴随CRC进展过程中,基因组稳定性失调带来的多次基因组不稳定性改变,以及产生的癌变后果。 APC 基因突变失活在高达85%的CRC中可见,与家族性腺瘤性息肉病密切相关。chr5q缺失造成的 APC 失活是最早观察到的基因组不稳定特征。除了在Wnt信号通路中的作用,突变的APC蛋白还可干扰微管末端与动粒的连接,并影响纺锤体检查点功能,造成有丝分裂异常,加剧CIN形成。当继发的CIN依次造成ch12p、ch18q和ch17P缺失时,造成 KRAS 原癌基因激活、 DCC/SMAD2/SMAD4 抑癌基因缺失、 TP53 抑癌基因缺陷。伴随ch20和ch8q重复及ch8p缺失造成的基因表达改变,细胞分裂增殖能力增强,CIN增多。 TP53 基因作为“基因组卫士”,守卫着DNA修复、细胞周期检查点、细胞凋亡等门户。 TP53 基因失活带来了灾难性的后果:具有大量的染色体异常和未修复DNA损伤的细胞被放过进入细胞周期分裂,造成基因组不稳定性的急剧上升,肿瘤由腺瘤转化成癌,癌变过程大大加快。在经历了ch8p缺失后,肿瘤细胞侵袭性增高,成为转移性癌。
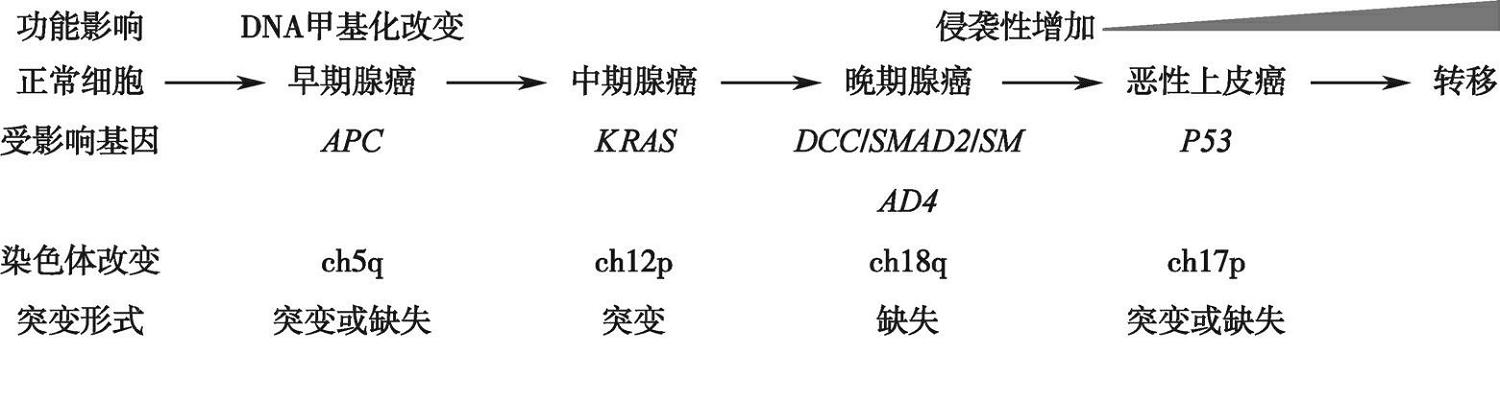
图3-3-2 基因组稳定性失调与结直肠癌变基因阶段性改变
基因组不稳定性加剧了癌变的过程,但同时,充分理解CRC中基因组不稳定性的发生机制,对CRC的预防、治疗和预后评估都具有重要意义。家族性的CRC前病变均具有其遗传学背景,因此具有家族史或在基因检测中发现易感基因的人群需有针对性地执行筛查和预防策略;不同的基因组不稳定性机制造成不同的MSI或CIN形式,而不同的机制缺陷也对不同的治疗策略易感。如PARP抑制剂对HR通路缺陷有协同致死效应,具有用于HR缺陷的CRC靶向治疗的潜力;热休克蛋白90抑制剂通过P53依赖的细胞死亡途径激活AMPK,对多倍体肿瘤颇具治疗功效;而具有MMR缺陷的CRC对5-氟尿嘧啶(5-fluorouracil,5-FU)治疗不敏感,对顺铂和卡铂敏感但却对其类似物不敏感。因此,充分评估肿瘤的基因组信息,对个体化治疗策略的制订非常重要;Carter等设计了一组包含 NXF1 、 P28 、 v - myb 、 RERG 、 STK15 等12个CIN相关调控基因的基因表达分析微阵列芯片用于预后评估,结果表明在乳腺癌、小细胞肺癌和CRC中,这些基因的特征性高表达均与肿瘤复发和不良预后密切相关,对于研发基于PCR的CRC临床风险预测和预后评估有极高参考价值。
传统观点的“突变体假说”认为在CRC中,因调控基因组稳定性的“守护基因”突变造成的基因组不稳定性在癌前病变中就已出现,并且驱动着肿瘤细胞发展过程中CIN的积累。在遗传性CRC病变中,该假说得到了充分验证。然而依靠蓬勃发展的高通量技术,研究却发现在散发病例中,除了 TP53 和 ATM ,并未有其他“守护基因”突变在早期癌前病变中高频出现。因此,近年来,有研究者提出了原癌基因介导DNA复制压力模型(oncogene-induced DNA replication stress model)。该假说认为:散发病例中,原癌基因表达导致DNA复制叉垮塌,从而造成DNA双链断裂和CIN,而“守护基因”的突变则出现于肿瘤发展的较晚阶段,并未参与早期的肿瘤发生。原癌基因导致的DNA复制压力模型解释了早期肿瘤中“守护基因”的低频突变,但原癌基因造成大量复制叉阻滞和垮塌的作用并未得到确切证实。也有研究者提出了“端粒侵蚀”等假说,但均欠完善。因此,散发病例中,CIN的调控和发生机制,仍待进一步研究。
(邵正萍)
CRC肿瘤细胞维持异常增殖的能力是其基本特点之一。正常结直肠上皮组织通过精确控制生长信号调控细胞增殖和分化周期,维持正常组织结构和功能。而CRC细胞通过多种方式获得持续增殖信号。癌细胞本身可以产生生长因子受体,通过同源受体表达引起自体增殖刺激。另外,支持肿瘤细胞的间质中,癌细胞可能发出信号刺激正常细胞,而正常的细胞则反馈活化各种生长因子,进而促进CRC细胞异常增殖。
正常结直肠黏膜上皮细胞看门基因如 APC 等出现突变导致其获得选择性生长优势逐渐形成腺瘤,腺瘤虽然生长缓慢,但一旦出现 KRAS 等基因突变,使GTP酶的负反馈调节失常导致细胞异常克隆性增殖,继而 PIK3CA 、 SMAD4 和 TP53 突变使其发展为具有侵袭性生长和转移潜能等恶性行为的CRC。这些驱动突变导致Wnt、EGFR/MAPK、PI3K-AKT和VEGF/VEGFR信号通路持续异常维持其异常增殖的潜能。
CRC细胞不仅具有诱导持续增殖刺激信号的能力,还具有能够逃逸抑癌基因依赖的增殖负调控程序。 Rb 和 TP53 是两个经典的抑癌基因,在调控细胞增殖、凋亡和衰老过程中发挥重要作用。 Rb 整合细胞内外多种信号决定细胞是否继续进入细胞的生长和分裂周期。CRC细胞通过表观遗传调控 Rb 表达或存在 Rb 信号调控通路功能性缺陷,导致细胞周期失控使癌细胞持续增殖。而 TP53 主要感应来自细胞内的压力和异常传输信号,如基因组损伤、细胞内核苷酸库水平、生长启动信号、乳糖或氧化水平异常, TP53 会阻断细胞周期进程,直至恢复正常化。当这些信号持续增强超出可修复程度, TP53 会诱导细胞的凋亡。CRC细胞中 TP53 表达缺失、突变及功能异常导致癌细胞异常修复及耐受凋亡进而表现为持续性增殖。以上以 Rb 和 TP53 为例说明CRC细胞具有逃逸抑癌基因的作用,而癌细胞中癌基因与抑癌基因相互作用的调控网络十分复杂,目前仍不清楚,需要进一步研究阐释。
CRC生长过程中,肿瘤细胞具有侵袭性生长特性,侵袭周围组织间隙和脉管系统,表现为局部浸润和远端转移。这些侵袭性生长的癌细胞其形状及与其他细胞和细胞外基质的黏附能力发生改变。E-钙黏着蛋白(E-cadherin)是关键的细胞间黏附分子,促进邻近上皮细胞黏附连接,形成上皮细胞层及维持上皮层的稳定。E-钙黏着蛋白的表达增加可以抵制肿瘤细胞侵袭和转移。而在CRC细胞中通常观察到E-钙黏着蛋白表达下调和偶见的突变失活。
CRC瘤细胞的运动和侵袭能力与EMT密切相关。肿瘤中EMT是指具有上皮样表型的肿瘤细胞转化为具有间质样表型的肿瘤细胞的过程,上皮性标志物如E-钙黏着蛋白、紧密连接蛋白1(zonula occludens,ZO1)等丢失,而间质性标志物如波形蛋白(vimentin)、N-钙黏着蛋白、纤维连接蛋白(fibronectin)和MMP等表达增加;肿瘤细胞在侵袭和转移的过程中,不同程度地短暂或永久激活EMT程序。而EMT过程的具体分子机制仍不清楚,但已知一些经典的EMT转录因子如 snail 、 slug 、 Twist 和 Zeb1/2 等参与了EMT的调控。这些转录调控因子在不同肿瘤细胞中或者肿瘤进展不同阶段以不同的组合表达,促进肿瘤细胞的侵袭和转移能力。CRC中肿瘤芽被认为是肿瘤细胞发生EMT的形态学表现。肿瘤芽是肿瘤浸润前缘单个肿瘤细胞或细胞数量少于5个的肿瘤细胞团,是一种处于非增殖、非凋亡,具有迁移和侵袭能力的特殊肿瘤细胞。肿瘤芽形态上为长梭形或纺锤形,细胞有伪足形成,上皮性标志物E-钙黏着蛋白表达下降,而间质性标志物波形蛋白和N-钙黏着蛋白等表达增加。
CRC细胞和间质细胞的相互作用同样促进肿瘤细胞的侵袭性生长和转移。例如存在于肿瘤间质中的间充质干细胞(mesenchymal stem cell,MSC)被发现能够分泌CCL5/RANTES响应癌细胞释放的信号,然后CCL5作用于肿瘤细胞,刺激肿瘤细胞的侵袭行为。肿瘤周围的巨噬细胞能够通过提供基质降解酶如金属蛋白酶和半胱氨酸蛋白酶促进肿瘤细胞局部浸润。此外,肿瘤相关巨噬细胞还能向肿瘤细胞提供表皮生长因子,而肿瘤细胞反过来用CSF-1刺激巨噬细胞,它们协同的相互作用能使肿瘤细胞更容易侵入循环系统和促进肿瘤细胞的转移扩散。
正常上皮细胞通过细胞与细胞之间的接触可以抑制细胞增殖,产生融合的细胞层。“接触抑制”机制的运行保证了体内正常组织稳态,然而在肿瘤发生的过程中该机制被破坏。但是,接触抑制及肿瘤如何摆脱接触抑制的分子机制仍不清楚。目前 NF2 作为肿瘤抑癌基因,编码膜突样蛋白(merlin),其结构与细胞骨架连接蛋白ERM家族[埃兹蛋白(ezrin)-根蛋白(radixin)-膜突蛋白(moesin)]相似,它通过将细胞表面黏附分子(如E-钙黏着蛋白)与跨膜酪氨酸激酶(如EGF受体)相互耦联而形成接触抑制。膜突样蛋白以这种方式强化了钙黏着蛋白介导的细胞与细胞间的黏附性。此外,膜突样蛋白还可以通过孤立生长因子受体而减弱有丝分裂的能力。另外,表皮极性蛋白LKB1参与组织表皮结构并维持组织完整性。当癌基因 MYC 表达上调时,LKB1能够拮抗 MYC 癌基因的促有丝分裂效应;而LKB1的表达抑制或失活导致表皮结构不稳定,细胞对 MYC 诱导的恶性转化更敏感。然而在CRC发生过程中,以上两种接触抑制机制的作用仍需进一步研究。
细胞凋亡是一种程序性细胞死亡,以凋亡小体形成为特点,不引起周围细胞损伤,也不引起周围组织炎症反应的单个细胞死亡。细胞凋亡途径分为由 BCL 家族控制的线粒体内源性细胞死亡途径,以及由死亡受体信号转导控制的外源性细胞死亡途径。
细胞凋亡与CRC的发病机制及其对化疗药物和放疗的耐药有关。正常结直肠黏膜稳态的维持依赖于隐窝底部干细胞增殖和表面上皮组织细胞凋亡之间的动态平衡,而这种平衡破坏后可能导致CRC的发生发展。CIN和MSI是CRC遗传不稳定的两个重要表现。细胞无法识别或修复遗传不稳定导致的错配,这些基因突变能够诱导细胞凋亡;而在一些CRC细胞中,凋亡的信号通路和效应通路失活则导致遗传突变的不断积累,使肿瘤进一步恶性进展,涉及APC/β联蛋白、RAS、TP53等信号通路异常。结直肠腺瘤发生过程出现 APC 突变,导致Wnt信号通路的异常激活和CIN,肠上皮细胞凋亡和增殖稳态被打破,出现异常增殖形成腺瘤。CRC细胞中 RAS/BRAF 突变活化MAPK信号通路导致肿瘤细胞抵抗凋亡能力增强,因此利用RAS的激酶抑制剂能够有效抑制其引起的凋亡抵抗。作为转录因子 TP53 参与许多细胞凋亡调控信号通路,调节了细胞凋亡与有丝分裂信号通路的相互作用,应答多种细胞应激。如 TP53 促进细胞周期蛋白依赖的激酶抑制基因 P21 上调,从而导致细胞周期阻滞,与 Bcl - 2 相互调控控制肿瘤细胞的凋亡。 TP53 还可以通过调控 IAP 和 survivin 等多种靶基因诱导细胞凋亡。
自噬,又称Ⅱ型程序性细胞死亡,是指细胞在饥饿和能量应激等状态下将自身的蛋白、细胞器和胞质进行包裹并形成囊泡,然后在溶酶体中消化降解的过程,包括大自噬、小自噬和分子伴侣介导的自噬。自噬是一种多步骤、多程序、高度进化及保守的过程,普遍存在于酵母菌、线虫、果蝇及哺乳动物等有机体中,对细胞维持其正常代谢和生存发挥着不可替代的作用。自噬的分子机制主要是以酵母为模型,利用遗传工程方法解析得到,在高等动物和真核细胞中高度保守。目前在酵母中已经发现20多种自噬相关基因,这些基因的功能在高等真核细胞中也高度保守。这些基因在5个重要阶段协调控制自噬这一复杂生物过程:①自噬泡的形成或核化;② Atg5-Atg12结合,并与Atg16L相互作用,形成Atg12-Atg5-Atg16L复合物,共聚到自噬泡上;③ LC3前体形成并加工成细胞质可溶性LC3-Ⅰ,然后修饰成膜结合形式的LC3-Ⅱ,与伸展和延伸的自噬泡融合;④随机或选择性捕获靶内容物,并将其降解;⑤与溶酶体融合形成自噬溶酶体。mTOR和Ⅲ级PI3K/Beclin1复合物等是调控自噬的重要信号转导通路。
自噬在肿瘤中具有“双刃剑”作用,不仅能抑制肿瘤的发生发展,还具有促进肿瘤进展的作用。一方面,自噬可以清除损伤的细胞器以及异常折叠的蛋白质,减少活性氧损伤从而保护正常肠上皮细胞并抑制其恶性转化。另一方面,自噬也能为肿瘤生长代谢提供丰富的营养物质。此外,自噬也可以影响癌细胞的化疗敏感性。在CRC中,自噬对CRC的化疗敏感性既有促进也有抑制作用,例如PCDH17可以通过诱导自噬增强CRC对5-FU的敏感性;而miR-22和IL-6等诱导的自噬导致CRC细胞对化疗的抵抗。自噬这种双重作用可能与诱导的自噬类型有关,但目前仍不清楚,需要进一步研究。
细胞焦亡是应对感染时激活的一种程序性细胞死亡,包括胱天蛋白酶(caspase)1依赖的经典焦亡和胱天蛋白酶4/5/11依赖的非经典焦亡。病原体的产物如脂多糖可促进炎性小体的形成激活胱天蛋白酶1。同时脂多糖也可以直接激活小鼠胱天蛋白酶11(人类同源基因为胱天蛋白酶4和5)。然后,胱天蛋白酶1和11都能剪切gasdermin D(GSDMD),使其N端片段在细胞膜上聚集打孔,破坏膜完整性导致细胞裂解。此外,GSDM蛋白家族的其他成员通过类似的机制参与细胞焦亡:gasdermin E由激活的胱天蛋白酶3或颗粒酶B剪切激活;gasdermin B由颗粒酶A剪切激活;gasdermin C由激活的胱天蛋白酶8剪切激活。与凋亡不伴随炎症反应不同,细胞焦亡可以释放出大量由胱天蛋白酶1剪切成熟的炎性物质IL-1β和IL-18激活天然免疫。
焦亡在肿瘤中发挥复杂的作用,焦亡可以直接介导肿瘤细胞死亡,同时也可以激活免疫反应,清除体内的肿瘤细胞。焦亡相关基因如 Nlrp3 或 caspase 1 缺失的小鼠更容易发生葡聚糖硫酸钠诱导的结肠炎和CRC。然而,也有学者提出细胞焦亡可能与自噬类似扮演“双刃剑”角色。GSDME介导的细胞焦亡通过释放HMGB1激活ERK1/2途径,促进肿瘤细胞增殖,继而促进CRC的发生发展。
铁死亡是一种铁依赖性的、非凋亡性的程序性细胞死亡形式,在形态、生化和分子机制与细胞凋亡、坏死、焦亡和自噬不同。其实质是细胞内脂质过氧化物代谢障碍。当亚铁离子催化作用产生的活性氧增加,当细胞内合成含多不饱和脂肪酸的磷脂增加,或者当细胞抗氧化能力减弱,使细胞内氧化还原失衡,含多不饱和脂肪酸的磷脂与活性氧反应产生的脂质过氧化物堆积,最终诱导细胞膜损伤,导致细胞死亡。由于该过程依赖二价铁离子,所以被称为铁死亡。形态学上,铁死亡主要表现为细胞体积变小,线粒体体积减小,线粒体膜密度增加,线粒体嵴减少或消失、破裂,细胞核大小正常且保持完整。铁死亡主要有内源性和外源性两种途径。外源性途径是通过抑制细胞膜上的胱氨酸/谷氨酸反向转运体或激活转铁蛋白与乳铁蛋白实现的,内源性途径则直接抑制细胞内抗氧化酶如GPX4活性从而诱导铁死亡发生。
铁死亡与肿瘤的发生发展密切相关。铁死亡同样具有促进和抑制肿瘤的双重作用。小分子化合物(如抑酯酶素和RSL3)、已被批准的药物(如索拉非尼、柳氮磺吡啶、他汀类和青蒿素)、电离辐射和细胞因子(如IFNγ和TGF-β1)可诱导铁死亡和抑制肿瘤生长。然而,铁死亡可在肿瘤微环境中触发炎症相关的免疫抑制,从而有利于肿瘤的生长。在肿瘤转移方面,血液中的高氧化应激环境可能会诱导肿瘤细胞的铁死亡从而抑制其血行转移,而淋巴结环境则能保护肿瘤细胞免于铁死亡。此外,铁死亡会影响化疗、放疗和免疫治疗的疗效。诱导铁死亡可以提高CRC细胞对西妥昔单抗的敏感性,抑制肿瘤细胞的转移。目前铁死亡在肿瘤中的研究处于刚刚起步阶段,仍需深入研究其在肿瘤中的作用和分子机制。
细胞衰老是指老化或受损的细胞发生持续性细胞周期阻滞,增殖能力降低的细胞状态。细胞衰老是一种多特征且高异质性的细胞状态,其表征包括但不局限于持续性细胞周期阻滞、溶酶体活性增强、抗凋亡刺激、细胞代谢异常、持续性DNA损伤及衰老相关分泌表型。
端粒被称为细胞有丝分裂的分子时钟,端粒的长度能够反映细胞分裂的程度和潜力,细胞有丝分裂过程中,由于DNA的单向复制特征,复制的DNA末端会出现“不完全复制”现象,导致端粒DNA在细胞有丝分裂过程中不断缩短,当端粒DNA长度缩短到一定“阈值”便能够触发细胞DNA损伤应答机制,DNA损伤相关激酶ATM、ATR、CHK1和CHK2进一步磷酸化P53蛋白,磷酸化的P53蛋白激活周期蛋白依赖性激酶抑制因子P21的表达,P21通过结合周期蛋白依赖性激酶2(cyclin-dependent kinase 2,CDK2),进而抑制细胞DNA复制,造成细胞出现“复制性衰老”现象。
衰老对于大多数物种而言是一种退行性病理状态,常伴随着组织和细胞功能的衰退。但是,在脊椎动物中,衰老能够促进增生性病变的发生,肿瘤与其他衰老性疾病特征相同,大部分类型的肿瘤发病率随年龄而增加,细胞能够通过衰老获得新的特征和功能,与肿瘤的发生发展密切相关。而肿瘤细胞衰老被认为是一种抗肿瘤机制,细胞衰老能够抑制肿瘤细胞的异常增殖。诱导肿瘤细胞衰老不但可以抑制肿瘤的恶性进展,而且能够通过激活衰老细胞的SASP进而诱导肿瘤的免疫杀伤。但有研究显示,P53介导的肿瘤细胞衰老能够降低肿瘤细胞的化疗敏感性和促进肿瘤的复发;化疗药物诱导的肿瘤细胞衰老却能够增强肿瘤细胞干性,促进肿瘤的发生和发展。
CRC细胞常伴有端粒酶表达激活的现象,端粒酶及端粒酶逆转录酶在结直肠腺瘤到腺癌转化过程中表达水平升高,其中myc及Wnt信号通路能够促进端粒酶基因的转录。此外,CRC细胞可由P53/P21信号通路介导其细胞衰老,MDM2蛋白表达增多能够抑制P53蛋白的乙酰化,进而阻止CRC细胞衰老进程,减少衰老相关异染色质点的形成。另外有研究表明,CRC中TRIB2和LRH-1蛋白表达增高,能够显著抑制P21蛋白的表达进而阻止CRC细胞的衰老。CRC细胞中CK1-α表达降低能够促进衰老CRC细胞分泌SASP,引发局部炎症反应,促进肿瘤的发展及转移。细胞衰老在肿瘤中的作用机制目前仍不清楚,其在肿瘤领域的应用仍需大量研究。
(张红河)
异质性是几乎所有晚期癌症的基本特征之一,相关研究证实肿瘤存在广泛的异质性,或者存在具有不同分化状态的肿瘤细胞。这些细胞不仅在生长增殖、侵袭转移以及对治疗反应性等方面有所不同,而且与其周围基质细胞特别是免疫细胞的相互作用等方面也存在巨大差异。对此现象的解释是多方面的,包括不同细胞克隆间的遗传和表观遗传变异,以及代谢和微环境的影响等,而肿瘤干细胞(cancer stem cell,CSC)理论认为,肿瘤中存在一个特定的细胞亚群,具有自我更新、增殖、多向分化潜能,与癌症转移、复发或肿瘤药物抗性密切相关,该细胞亚群被定义为CSC。
事实上,CSC的概念起源于20世纪90年代对白血病的研究,发现白血病具有与正常造血系统类似的等级系统,其肿瘤细胞是从一小部分干细胞发展而来,这些干细胞既能自我更新,又能产生一系列分化程度更高的细胞。通过将造血干细胞的定义进行扩展,从而建立了体内鉴定CSC成瘤能力和干性的金标准,即将分离的肿瘤细胞移植给受体小鼠,极少量的细胞(最少至100个)就可以形成反映原始患者肿瘤特征的移植瘤,同时该肿瘤细胞具有无限自我更新潜能,则认为其具有肿瘤干性。
近年来,对CRC干细胞(colorectal cancer stem cell,cCSC)功能特征和分子调控机制进行了大量研究。研究表明cCSC已经不是最初认为的具有明确表型和分子特征的细胞亚群,而是受遗传、表观遗传和微环境共同影响而动态改变的细胞亚群。因此,阐明cCSC驱动CRC进展的机制可能为临床提供新的干预途径,从而最终改善治疗效果。
小肠上皮由底部隐窝结构及表面绒毛组成,两者间以隐窝-绒毛移行细胞构成,是体内更新速度较快、再生能力较强的组织之一,也是干细胞研究的经典模型,每3~5天更新一次,而新肠上皮细胞是由肠道干细胞(intestinal stem cell,ISC)不断增殖分化产生的。尽管cCSC不一定直接起源于正常ISC,但由于正常和癌变的干细胞往往会依赖于相同的信号通路,因此研究调控ISC的分子机制将有助于加深对cCSC的了解。目前小肠在功能上分为不同的2种肠道干细胞亚群:分裂活跃的隐窝基部柱状细胞高表达LGR5和处于静息状态的“+4”位细胞,即储备干细胞普遍表达BMI1、HOPX、TERT和LRIG1,这两群ISC都具有自我更新和进一步分化出所有肠上皮细胞的能力。
最初研究认为这两种干细胞亚群代表具有不同功能活性的亚群:增殖活跃的LGR5 + 柱状干细胞被认为是维持肠稳态的原因,而静态的BMI1 + 储备干细胞则被视为能够再生LGR5 + 亚群的储备干细胞池。然而,后续研究发现LGR5 + 柱状干细胞也可以表达“+4”标记(如BMI1等),这就模糊了两个ISC亚群之间的界限。另外,运用谱系示踪技术特异性消融ISC后,肠内分泌细胞、DLL1 + 分泌祖细胞、标签滞留细胞、KRT19 + 上层隐窝中的祖细胞和帕内特细胞等祖细胞甚至高度分化的细胞将在损伤刺激作用下逆向去分化,转变成具有干性的ISC。这些研究表明小肠上皮中存在的多种祖细胞具有较高的可塑性,可以在应激情况下逆向去分化以补充ISC。
与小肠相比,结肠在隐窝结构和细胞组成方面存在差异:如结肠隐窝不会在黏膜表面形成绒毛,也不包含帕内特细胞、“+4”位细胞或BMI1 + 细胞。已有的研究表明,结肠干细胞主要为表达LGR5 + 或EphB high 的细胞亚群,当被植入小鼠后具有重建整个结肠的自我更新能力。另外,后续研究还发现了其他的结肠干细胞亚群,如高表达LRIG1或Notch信号的慢分裂干细胞以及表达DCLK1 + 肠道簇细胞的亚群。然而,与小肠相比,维持结肠干细胞干性或可塑性的分子机制研究还相对薄弱,特别是结肠内寄居着比小肠更多的细菌群落,其作用有待后续研究。
对于多数肿瘤而言,通常认为干细胞是肿瘤恶变的起始细胞,即组织中正常的干细胞获得了向肿瘤恶性转变所需的原始突变积累。在转基因小鼠模型,当特异性激活小肠隐窝底部LGR5 + 、CD133 + 或BMI1 + 正常ISC的Wnt信号通路时,可见腺瘤的快速生长。相比之下,在已分化的细胞中特异性地缺失APC(Wnt通路负向调控因子)只会偶尔导致腺瘤的发生,表明正常ISC比已分化细胞更容易发生恶性转化。虽然正常隐窝中的ISC以随机的方式进行互相替换,但致癌突变赋予了其突变克隆竞争优势,从而较少被野生型ISC替代。值得注意的是,尽管突变的ISC比野生型更有竞争优势,但其细胞命运并不是固定的,仍然有可能被野生型随机替代,从而使得突变积累变得更为复杂。另外,研究还发现在正常条件下 P53 突变的ISC并不比野生型更有优势,但肠炎的发生将使突变型ISC更有竞争优势。这些实验结果清楚地表明遗传和环境因素共同决定了ISC克隆的竞争能力,影响着CRC的早期发生。
近年来,正常ISC作为CRC起始细胞的观念开始受到挑战,在已分化的绒毛细胞中异常激活NF-κB信号通路(如肠炎或 KRAS 突变)后,这些细胞变得更易于癌化,但只激活绒毛细胞中的Wnt通路则不足以引发腺瘤。这些现象表明CRC的起始需要Wnt通路以外的其他信号(如NF-κB等)的共同参与,这些信号的存在扩大了CRC的起始细胞范围。因此,错构瘤性息肉病等肠炎人群中的CRC风险增加或许与此机制有关。此外,研究还表明已分化DCLK1 + 肠道簇细胞在 APC 基因缺失和葡聚糖硫酸钠诱导肠炎的共同作用下,也可以导致CRC的发生。综上所述,干细胞(如ISC等)、祖细胞或已高度分化的细胞(绒毛细胞等)都可能是CRC的起始细胞。
多年来,人们一直致力于寻找cCSC特异性的生物标志物,这将极大地促进针对CRC预防和治疗干预工具的研究。最初基于CD133表达高低,运用流式分选分离了人类cCSC干细胞亚群,并异种移植到免疫缺陷鼠,产生了与人源CRC类似的小鼠肿瘤。目前研究已经确定的cCSC分子标志物有EphB2 high 、EpCAM high /CD44 + /CD166 + 、ALDH + 、LGR5 + 和CD44v6 + 等,但这些标志物通常在正常ISC中也表达,限制了其作为治疗靶标的潜能。近年来,研究还发现DCLK1在cCSC中特异性地高表达,而不表达于ISC中,是潜在的cCSC治疗靶标。
尽管cCSC领域的研究取得了长足进展,但仍然面临着一些问题。首先,cCSC分子标志物的一致性还值得进一步研究,因为其表达本身是不稳定的,研究发现LGR5 + cCSC亚群和LGR5 - cCSC亚群可以共存于同一肿瘤,并在化疗时相互转化。此外,肿瘤微环境中的基质细胞也会产生各种细胞因子(如Wnt、TGF-β和R-spondin等)影响cCSC的自我更新能力,并可在应激损伤时促使祖细胞逆向去分化产生cCSC。因此,不仅表达特定分子标志物的cCSC的细胞占比可能会随肿瘤的进展阶段以及治疗方案的不同而动态变化,同时其表达的分子标志物类型和数量也不断发生改变。
越来越多的证据表明CSC具有通过基因表达和表观遗传学改变进行适应性状态改变或表型转换的能力,即可塑性。这个概念不仅包含cCSC在耐药性、不对称分裂和分化状态等方面获得不同表型的能力,也包含其他已分化的肿瘤细胞亚群在特定情况下逆分化成cCSC的潜能。早期研究表明,Lac-Z报告基因标记的CD133 + 和CD133 – 癌细胞都可导致免疫缺陷鼠中转移瘤的发生。此外,研究还发现LGR5 + cCSC在 APC Min/+ 小鼠辐照后的腺瘤复发过程不是必需的,而KRT19 + /LGR5 - 细胞亚群被发现才是导致腺瘤复发的重要因素。运用白喉毒素特异性地清除LGR5 + cCSC后,发现小鼠的肿瘤生长基本不受影响。此外,运用类器官和人源肿瘤异种移植(patient-derived tumor xenograft,PDX)模型,通过激活caspase 9特异性地清除LGR5 + cCSC从而抑制了肿瘤的生长;然而,当停止清除LGR5 + 细胞后,肿瘤的复发主要由KRT20 + /LGR5 - 细胞亚群驱动。以上研究清楚地表明表达相关分子标志物的cCSC并不是CRC发生的唯一驱动因素,其他的非干细胞亚群在损伤刺激作用下也可以逆分化成cCSC。
近年来,基于 CRISPR/Cas9 的无荧光标签标记技术和高通量测序技术的发展,通过分析细胞中随机引入的短序列/突变等遗传条形码来进行谱系示踪。运用此技术,研究发现来源于同一患者肿瘤的cCSC,尽管在遗传上表现出高度的均一性,但在耐药性、不对称分裂和分化状态等方面有诸多不同表型。最近研究还发现cCSC比其他非干细胞具有较高的核糖体活性和蛋白合成能力,但这些高活性基本与cCSC所带有特定的基因突变谱无关。此外,多年的研究也已证实驱动肿瘤发生的关键驱动突变大多发生在早期阶段,而在后续进展过程中仅有有限的功能突变发生。总之,在肿瘤的演进过程中,决定cCSC功能特性的关键因素并不是早期的遗传突变,而是DNA甲基化、组蛋白修饰和染色质可及性等表观遗传修饰。
肿瘤微环境中不同的基质细胞和细胞外基质共同组成了cCSC的干细胞巢,其组成随肿瘤的发展而不断动态变化,对cCSC细胞的命运决定和功能特征发挥至关重要的作用。运用PDX模型追踪表达GFP报告基因(由Wnt启动子驱动)的cCSC,研究发现驱动肿瘤进展的cCSC主要位于肿瘤的边缘,紧靠微环境中的基质细胞。后续研究也证实来自周围基质细胞的信号在调节肿瘤细胞中的Wnt激活水平上具有重要作用。此外,运用多色谱系示踪系统对来源于患者的cCSC进行更长时间的追踪观察,发现cCSC克隆的大小与其距离肿瘤边缘的物理位置有较大的相关性。这些研究表明cCSC在微环境中所处的细胞位置是肿瘤进展过程中克隆竞争的主要驱动因素。
实际上,处于肿瘤边缘处的肿瘤细胞时刻暴露于来源于其周围间质细胞的信号网络中(如Wnt、TGF-β和R-spondin等),这些细胞因子共同作用影响cCSC的自我更新能力。例如,Lenos等的研究发现基质细胞分泌的骨桥蛋白是肿瘤边缘cCSC克隆竞争性扩增的诱导因素。微环境中肿瘤相关的成纤维细胞旁分泌PGE2,与其周围肿瘤细胞上的PTGER4受体结合,调控Sca-1 + 肿瘤起始细胞的自我更新能力和肿瘤的发生。
除了微环境中间质细胞可以影响cCSC的干性潜能和肿瘤发生,癌变的cCSC也会影响正常ISC的自我更新能力,从而使微环境中肿瘤细胞和间质细胞的相互交流变得更为复杂。研究发现APC缺失的腺瘤细胞可以分泌NOTUM(Wnt通路拮抗剂),从而抑制同一隐窝或相邻隐窝内正常ISC的干细胞潜能。此外,通过正常和癌变干细胞的独立示踪模型,研究发现受癌基因激活驱动的肿瘤细胞不仅自我分泌抑制性细胞因子从而诱导周围正常ISC的调亡和分化,也会促使其周围其他间质细胞分泌抑制性细胞因子。总之,在肿瘤的演进过程中,受癌基因突变驱动的肿瘤细胞将通过旁分泌影响其他间质细胞,重塑形成对己有利的微环境,从而维持cCSC的自我更新和分化。
近年来,所有关于cCSC的研究都汇集到一点:如何重新定义cCSC并靶向cCSC进行肿瘤治疗。学者们逐渐认识到cCSC并非是肿瘤中具有固有特征的特定细胞亚群,而是随肿瘤的演进不断发生动态改变,在不同情况下由不同的细胞亚群承担。这种可塑性除受肿瘤本身和基质等微环境的影响外,同时还有一系列其他未知因素的参与。此外,影响cCSC干性和克隆竞争的决定因素也会随肠癌的不同发展阶段而有所不同。总之,cCSC复杂多变的异质性和可塑性也向人们提出了巨大的挑战,这可能正是目前靶向CSC进行肿瘤治疗失败的主要原因,还有待后续研究。
(刘云华)
肿瘤是一种旺盛增殖的组织,为满足细胞快速增长和分裂的需要,其细胞内物质代谢发生了一系列特征性改变,表现为组成细胞基本结构的物质如蛋白质、脂类和核酸的合成十分旺盛,相反,氨基酸和核苷酸的分解代谢则显著降低,导致合成代谢和分解代谢的平衡失调。
葡萄糖是细胞最重要的能量物质来源,葡萄糖在细胞内的经典代谢通路如图3-6-1所示,葡萄糖代谢有三种关键酶:己糖激酶(hexokinase,HK)、磷酸果糖激酶1(phosphofructokinase 1,PFK1)、丙酮酸激酶(pyruvate kinase,PK)。首先,葡萄糖被Na + 依赖的葡萄糖转运体从血流中主动运输进入细胞,随即被HK磷酸化生成6-磷酸葡萄糖(glucose-6-phosphate,G6P)。G6P作为关键代谢中间产物,位于两种主要代谢途径——糖酵解途径和戊糖磷酸途径(pentose phosphate pathway,ppp)的分支点。当细胞中NADP + /NADPH比率增高时,G6P倾向于进入ppp,被6-磷酸葡萄糖脱氢酶催化脱氢,生成5-磷酸核糖和NADPH。如果细胞中ATP水平降低,G6P将被磷酸葡萄糖异构酶变构为6-磷酸果糖(fructose-6-phosphate,F6P),倾向于进入糖酵解途径。随着ATP能量被消耗,F6P受PFK1催化生成1,6-二磷酸果糖(fructose-1,6-bisphosphate,F1,6-BP),该步骤是糖酵解过程中的关键调控步骤。

图3-6-1 糖代谢经典途径
醛缩酶进一步将F1,6-BP分解生成两分子内糖磷酸二羟丙酮(dihydroxyacetone-P)和3-磷酸甘油醛(glyceraldehyde-3-phosphate,GA3P)。前者在磷酸丙糖异构酶的催化下转化为GA3P,继续进行酵解。随后,GA3P脱氢氧化生成1,3-二磷酸甘油酸(1,3-bisphosphoglycerate,1,3-BPG),此步由甘油醛脱氢酶催化,并生成NADH。1,3-BPG由磷酸甘油酸激酶(phosphoglycerate kinase,PGK)催化生成3-磷酸甘油酸(3-phosphoglycerate,3-PG),3-PG被磷酸甘油酸转位酶(phosphoglycerate mutase,PGM)转变为2-磷酸甘油酸,进一步由烯醇化酶催化生成磷酸烯醇式丙酮酸(phosphoenolpyruvate,PEP)。
最后,PEP被PK催化生成丙酮酸(pyruvate,Pyr)。如果丙酮酸不进入线粒体途径被利用,将被细胞质中的乳酸脱氢酶(lactate dehydrogenase,LDH)转变为乳酸。乳酸则通过分布于胞膜的单羧基转运体分泌至胞外,进入血液循环,到达肝脏后通过糖异生作用,转变成肝糖原或血糖,形成乳酸循环。
总体来说,细胞产生ATP的方式有两种:细胞质中不依赖于氧的糖酵解途径、线粒体中依赖于氧的氧化磷酸化途径(oxygendependent oxidative phosphorylation,OXPHOS)。糖酵解途径是从葡萄糖开始分解生成丙酮酸的过程,生物在无氧条件下,能够通过糖酵解产生ATP与还原型辅酶NADH。NADH在糖酵解、三羧酸循环、β氧化等多个代谢过程均有产生,它含有极高电极电势的电子,在氧化时将释放出大量能量。在氧化磷酸化途径中,线粒体内膜上的酶类通过氧化NADH释放能量,泵送质子穿过线粒体内膜,从而产生跨膜电化学梯度,ATP合酶利用势能产生ATP。
两种途径在ATP生产过程中的占比由细胞供氧情况和线粒体功能状态决定。在供氧充分、线粒体功能正常时,丙酮酸进入线粒体,生成乙酰辅酶A,通过三羧酸循环氧化,再通过电子传递链产生大量ATP。正常生理条件下,细胞所需的绝大多数ATP通过线粒体氧化磷酸化途径产生,而肿瘤细胞的糖代谢则发生明显改变。
在正常组织中,约90%的ATP来源于线粒体的氧化磷酸化途径,仅10%来源于糖酵解途径。与之相反,恶性肿瘤细胞的糖酵解明显增强,即使在氧气供应充足的条件下,大多数来自糖酵解的丙酮酸被引导离开线粒体,通过乳酸脱氢酶作用产生乳酸,该现象称为有氧糖酵解或瓦尔堡效应(Warburg effect)。
在CRC组织中,瓦尔堡效应的产生有多方面因素,其中,癌基因 MYC 、抑癌基因 P53 、转录因子HIF-1和AKT激酶等对于促成肿瘤瓦尔堡效应的细胞代谢调控至关重要。癌基因 MYC 的激活将上调糖酵解相关分子表达,如葡萄糖转运蛋白1、HK和LDH,使糖酵解代谢增强; RAS 等癌基因信号、肿瘤微环境中的 HIF - 1 可能作用于糖酵解相关基因,从而增加肿瘤细胞糖酵解流量。 HIF - 1 可以激活 PDK1 基因表达,进而抑制PDH,限制丙酮酸进入线粒体产生ATP。AKT上调葡萄糖转运蛋白1水平,增强HK与线粒体结合。
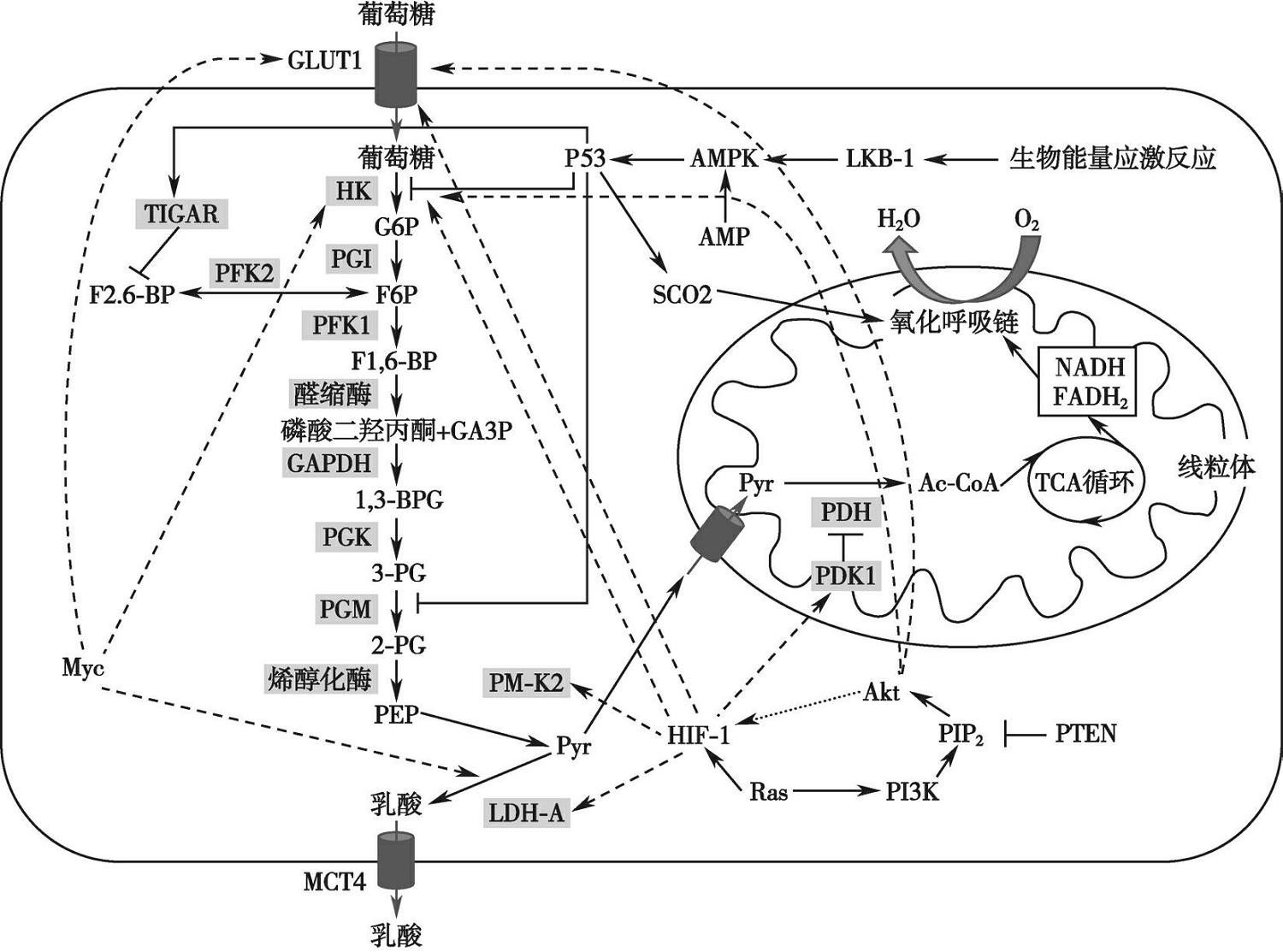
图3-6-2 瓦尔堡效应影响下的代谢调控途径
肿瘤抑制因子P53则发挥相反的抑制作用,通过调控HKⅡ、PGM和TIGAR表达抑制糖酵解。TIGAR是磷酸果糖激酶PFK2的一个亚型,能够降低细胞内2,6-二磷酸果糖水平,而后者为糖酵解关键限速酶PFK1的主要变构激活剂,因此P53能够通过激活TIGAR,抑制PFK1的激活,减少糖酵解代谢。P53还能够通过转录调控细胞色素C氧化酶2,促进线粒体功能的行使。AMP依赖激酶(AMP-dependent kinase,AMPK)对糖酵解的调控功能也部分通过激活P53实现。P53功能缺失的肿瘤细胞糖酵解功能将增加,线粒体呼吸作用减弱,导致代谢重编程的发生。
瓦尔堡效应支持肿瘤细胞不受控增殖的生物合成需求。在糖酵解明显增强,ATP产生效率降低的情况下,葡萄糖代谢产生的大量中间产物成为支持肿瘤细胞合成代谢的碳源物质。肿瘤细胞利用这些碳源物质进行核苷酸、脂质和蛋白质的快速合成,以支持其迅速生长和增殖。
瓦尔堡效应为多细胞环境中的癌细胞生长提供优势。瓦尔堡效应的特点是每分子ATP的产酸量高,乳酸分泌、葡萄糖代谢升高会降低肿瘤微环境pH。现代癌症进展假说认为,肿瘤细胞拥有比正常细胞更强的排酸能力,保护其细胞内结构免受酸累积的破坏。因此,pH较低的胞外环境能够促进肿瘤细胞生长发育,而不利于正常细胞生存。同时,糖酵解(即产酸)强度与肿瘤侵袭性成正相关关系,肿瘤分泌的乳酸能够促进M2组织相关巨噬细胞极化,推动肿瘤免疫逃逸的发生。
研究表明,对 KRAS 突变的结肠癌细胞系,葡萄糖代谢抑制剂如HIF-1α抑制剂、MCT1抑制剂和葡萄糖转运蛋白1抑制剂均有较好的抑制效果。进一步地,糖酵解抑制剂可以与血管生成抑制剂联合用药,改善CRC细胞对血管生成抑制剂的耐药情况。糖代谢作为肿瘤抑制靶点,可以促进肿瘤细胞的凋亡,抑制其生长与传播。
甘油三酯是体内储量最大和产能最多的能源物质。甘油三酯的分解代谢从脂肪动员开始。储存在脂肪细胞中的脂肪在脂肪组织甘油三酯脂肪酶、激素敏感性脂肪酶和脂滴包被蛋白-1等脂肪酶的作用及激素调节下,可逐步水解并释放出游离脂肪酸和甘油供其他组织氧化利用。在细胞中,甘油可被甘油激酶催化生成3-磷酸甘油,再脱氢生成磷酸二氢丙酮,进入糖分解或糖异生途径被利用;而脂肪酸也可通过各种氧化分解途径为细胞供能。
β氧化途径是人体细胞内脂肪酸分解代谢的主要途径。在脂酰CoA合成酶的作用下,脂肪酸先被活化生成脂酰CoA,接着经过肉碱穿梭转运进入线粒体中进行β氧化。在β氧化的过程中,转运入线粒体的脂酰CoA将进行“脱氢、加水、再脱氢、硫解”的4步反应循环,每次生成1分子乙酰CoA和缩短二碳的脂酰CoA,后者继续循环直至全部生成乙酰CoA。脱氢生成的FADH 2 、NADH经呼吸链氧化,与ADP磷酸化耦联,产生ATP供细胞利用;生成的乙酰CoA主要在线粒体通过三羧酸循环彻底氧化。
在肝脏内,脂肪酸可氧化分解成乙酰乙酸、β-羟基丁酸和丙酮,这类产物称为酮体,可在肝外组织的线粒体中被利用。葡萄糖供应充足的情况下,脂肪合成代谢增强,分解代谢减少,酮体生成减少;反之则酮体生成增加。
肝、脂肪组织及小肠是甘油三酯合成的主要场所。甘油和脂肪酸作为合成甘油三酯的原料,主要来源于食物及葡萄糖分解代谢的中间产物。脂肪酸先活化生成脂酰CoA,然后于小肠黏膜细胞通过甘油一酯途径,肝脏和脂肪组织通过甘油二酯途径合成甘油三酯,后者主要原料来源于葡萄糖分解代谢的中间产物。
内源性脂肪酸的合成需首先由乙酰CoA在脂肪酸合酶复合体催化下合成软脂酸。乙酰CoA羧化生成丙二酸单酰CoA,在内质网或线粒体内经过7次“缩合、还原、脱水、再还原”循环合成软脂酸。这一反应与脂肪酸β氧化的逆反应相似。
磷脂是一类含有磷酸的脂类,是组成生物体膜的主要成分。机体中主要含有两大类磷脂:由甘油构成的磷脂,称为甘油磷脂;由神经鞘氨醇构成的磷脂,称为鞘磷脂。
甘油磷脂合成所用的甘油、脂肪酸主要由糖代谢转化而来,甘油碳骨架C2上连接的多不饱和脂肪酸常需靠食物供给,合成除ATP、GTP提供能量外,还需要CTP作为活化因子。甘油磷脂分子内的三种酯键可分别被相应的磷脂酶催化水解,生成不同的降解产物。
鞘磷脂是神经鞘磷脂合成的重要中间产物。软脂酰CoA、丝氨酸在内质网合成酶系的催化下先生成鞘氨醇,再与脂酰CoA缩合生成神经酰胺,最后与CDP-胆碱结合生成神经鞘磷脂。在神经鞘磷脂酶的作用下,神经鞘磷脂又可分解为磷酸胆碱和神经酰胺。
细胞内胆固醇合成的主要酶系存在于细胞质和内质网。胆固醇的合成主要有三个步骤:3分子乙酰CoA先连续缩合生成羟甲基戊二酸单酰辅酶A(hydroxymethylglutaryl CoA,HMG-CoA),再经HMGCoA还原酶催化生成甲羟戊酸(mevalonate,MVA);MVA活化生成2碳焦磷酸化合物(异戊烯醇焦磷酸酯(IPP)和2,6-二苯基苯酚(DPP)后,连续缩合为15碳法尼基焦磷酸(farnesyl pyrophosphate,FPP),2分子FPP缩合成30碳的鲨烯;鲨烯环化为羊毛固醇,再经多步反应生成27碳的胆固醇。在肝内转化为胆汁酸是胆固醇的主要去路。此外,胆固醇还可转变为类固醇激素和维生素D 3 。
肿瘤细胞可通过上调脂肪酸转运载体和脂质分子伴侣,如脂肪酸转运蛋白、脂肪酸结合蛋白,增加游离脂肪酸的摄取,提高脂类生物利用度,使肿瘤细胞获得足够的能量来增殖。
与正常细胞相比,肿瘤细胞中的脂肪酸合成代谢常异常激活,包括脂肪酸的从头合成与甘油三酯合成。有研究发现,肿瘤细胞中(mTOR)信号通路的激活可诱导脂肪酸合成酶过表达,使细胞内脂肪酸合成增多,进而引起脂肪合成增多,为肿瘤细胞生长、增殖提供充足的能量。与此同时,肿瘤细胞脂肪酸氧化分解也常异常活化。脂肪酸氧化增强比丙酮酸的氧化产生ATP的效率更高,因而有利于适应肿瘤细胞的高耗能状态,从而促进肿瘤生长。
肿瘤细胞中脂质成分的异常代谢会影响细胞凋亡、增殖、迁移、侵袭等多种生物学行为,进而影响肿瘤的发生发展和患者的预后水平。有研究表明,肿瘤细胞中胆固醇的合成水平较正常细胞升高。而细胞质中的溶酶体胆固醇可通过肠道胆固醇转运关键蛋白信号复合物激活mTORC1,进而导致癌细胞增殖、侵袭和转移增加。此外,鞘磷脂在CRC细胞内的水平也发生了改变,具体表现为1-磷酸鞘氨醇/神经酰胺比率的变化。而目前有文献指出,神经酰胺增加可导致肿瘤细胞的凋亡和自噬激活,抑制细胞增殖和肿瘤发展。
异常脂代谢可影响肿瘤微环境中的免疫细胞。研究表明,在CRC中,巨噬细胞、树突状细胞、淋巴细胞、自然杀伤细胞都存在脂肪酸合成及氧化分解增加的现象;组织常驻记忆T细胞和调节性T细胞(regulatory T cell,Treg细胞)表现出对外源脂肪酸合酶(Fatty acid synthase,FAs)摄取效应增强并在线粒体中氧化分解以产生足够的能量。这些证据均可说明肿瘤细胞和肿瘤微环境中的免疫细胞相互作用可导致双方脂代谢重编程,进而影响肿瘤进展。
癌症相关成纤维细胞中的脂代谢重编程可以促进CRC细胞的迁移。研究发现,成纤维细胞和脂肪细胞分泌的脂肪酸可被CRC细胞吸收用于合成其他脂质,进而促进CRC细胞的迁移和侵袭表型。
来自食物蛋白质的消化吸收、体内组织蛋白质的降解以及少量自身合成的非必需氨基酸构成了人体氨基酸代谢库。这些氨基酸的主要代谢去路有:作为合成组织蛋白质的原料;脱氨生成α-酮酸代谢供能;代谢转化为其他含氮化合物及化学基团,如γ-氨基丁酸等生物活性物质及嘌呤、嘧啶、一碳单位等。
氨基酸脱氨基生成相应的氨及α-酮酸,是氨基酸分解代谢的主要途径。在转氨酶及其辅酶磷酸吡哆醛(维生素B 6 )的作用下,某一氨基酸的α-氨基转移至另一种α-酮酸的酮基上,生成相应的氨基酸,原来的氨基酸则转变成α-酮酸,可用于人体非必需氨基酸合成、转变为糖或酮体或氧化供能。转氨基作用只是将氨基酸分子中的氨基转移给α-酮酸,并未实现真正的脱氨基。L-谷氨酸是哺乳动物组织内唯一能以相当高速率进行氧化脱氨反应的氨基酸。在肝、肾、脑中,L-谷氨酸可在谷氨酸脱氢酶的催化下脱去氨基,生成α-酮戊二酸和氨。转氨基作用与L-谷氨酸的氧化脱氨基作用耦联进行,称为联合脱氨作用。
各种氨基酸经过代谢转化,可产生或转化为不同的化合物(表3-6-1)。
表3-6-1 个别氨基酸的代谢方式、产物和主要功能
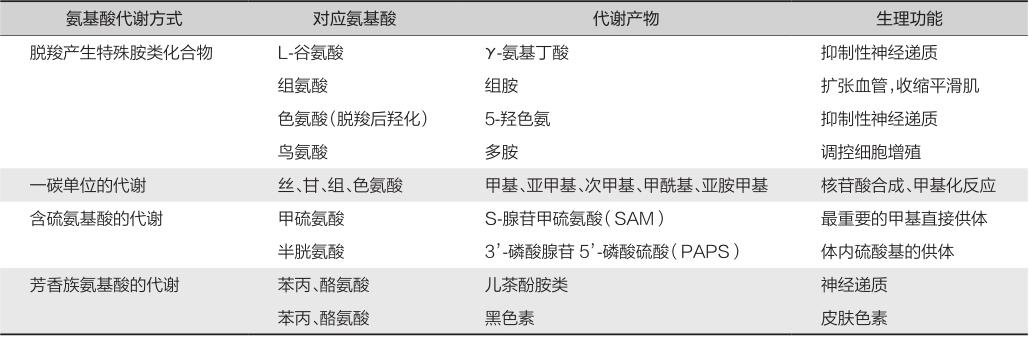
丝氨酸、甘氨酸、组氨酸和色氨酸的分解代谢可以产生一碳单位。一碳单位是合成嘌呤、嘧啶核苷酸乃至核酸的原料,与细胞增殖有着密切的联系。
谷氨酰胺在细胞能量生成、大分子合成及信号转导等功能中发挥着重要作用。除了代谢产生能量,谷氨酰胺还是合成许多生物重要分子的前体,并提供还原型辅酶Ⅱ(NADPH)和谷胱甘肽来维持氧化还原平衡,因此对肿瘤细胞的生长和增殖起着十分关键的作用。
肿瘤细胞可通过上调谷氨酰胺转运体、调节谷氨酰胺代谢相关酶促反应等途径增强对其的代谢反应,从而促进肿瘤进展。研究表明,许多肿瘤细胞表现出“谷氨酰胺成瘾”的特征,而一旦通过各种手段阻断细胞对谷氨酰胺的摄取及利用,肿瘤的生长和发展将会停滞。
一碳单位的代谢与可以调控核酸、蛋白质和脂质的合成,维持细胞的氧化还原内稳态及表观遗传的稳定性,进而影响肿瘤的发生发展。叶酸循环是一碳单位的代谢核心反应,受阻时细胞核酸合成将会发生障碍,由此可以抑制肿瘤细胞的生长增殖,由此研发了CRC抗甲基化化疗药物5-FU。甲硫氨酸循环能够影响DNA的甲基化程度,与CRC密切相关,DNA甲基转移酶抑制剂可作为治疗CRC的新方式。生成一碳单位的原料氨基酸如丝氨酸、甘氨酸等,针对它们的代谢研究也可能为CRC相关机制及治疗手段的探索带来新的思路。
(卓巍)
表观遗传学是指研究基因表达或蛋白表达的改变、不涉及DNA序列变化,但又可以通过细胞分裂稳定遗传现象的遗传学分支领域,包括DNA甲基化、组蛋白修饰及染色质重塑等。1942年,Waddington首先提出了“表观遗传学”这一概念,用以定义基因型未发生改变而表型改变的现象,以解释发育的各个方面,阐释了生物体从基因到基因表型之间存在一种控制机制。1987年,Holiday发现表观遗传研究包含有丝分裂和减数分裂在细胞和个体世代间传递,而非DNA序列的改变。1995年,约翰·霍普金斯大学(Johns Hopkins University)医学院的Stephen Baylin发现多种人体肿瘤中抑癌基因周期蛋白依赖性激酶抑制因子2A(cyclin dependent kinase inhibitor 2A,CDKN2A)呈高甲基化,而用去甲基化抑制剂处理,能保持 CDKN2 基因的活性,提示甲基化能使抑癌基因活性沉默。
结直肠肿瘤的发生发展是一个多因素、多阶段的复杂过程,涉及一系列遗传学和表观遗传学累积的改变。近年来,随着人们对表观遗传学认识的深入,尤其是DNA甲基转移酶抑制物,组蛋白乙酰化抑制剂等在治疗肿瘤患者的成功临床应用,表观遗传学逐渐成为肿瘤研究的热点。
DNA甲基化是调节基因表达的最普遍的表观遗传修饰之一,由DNA甲基化转移酶催化S-腺苷甲硫氨酸作为甲基供体,在基因的5′-CG-3′二核苷酸的胞嘧啶的第五位碳原子上共价结合一个甲基基团,形成5-甲基胞嘧啶。CpG岛是指CpG序列的密度比平均密度高10~20倍,GC含量大于50%,长度为200~2 000bp的区域。哺乳动物细胞中,CpG二核苷酸在人类基因组中约占10%,包括散CpG和CpG岛。其中,70%~90%的散在CpG呈高甲基化状态,而大部分CpG岛中的CpG呈低甲基化状态。CpG岛主要位于结构基因的启动子和第一外显子区域,60%~70%的基因启动子含有CpG岛。病理状态下,DNA甲基化模式的改变包括DNA高甲基化和DNA低甲基化(图3-7-1)。
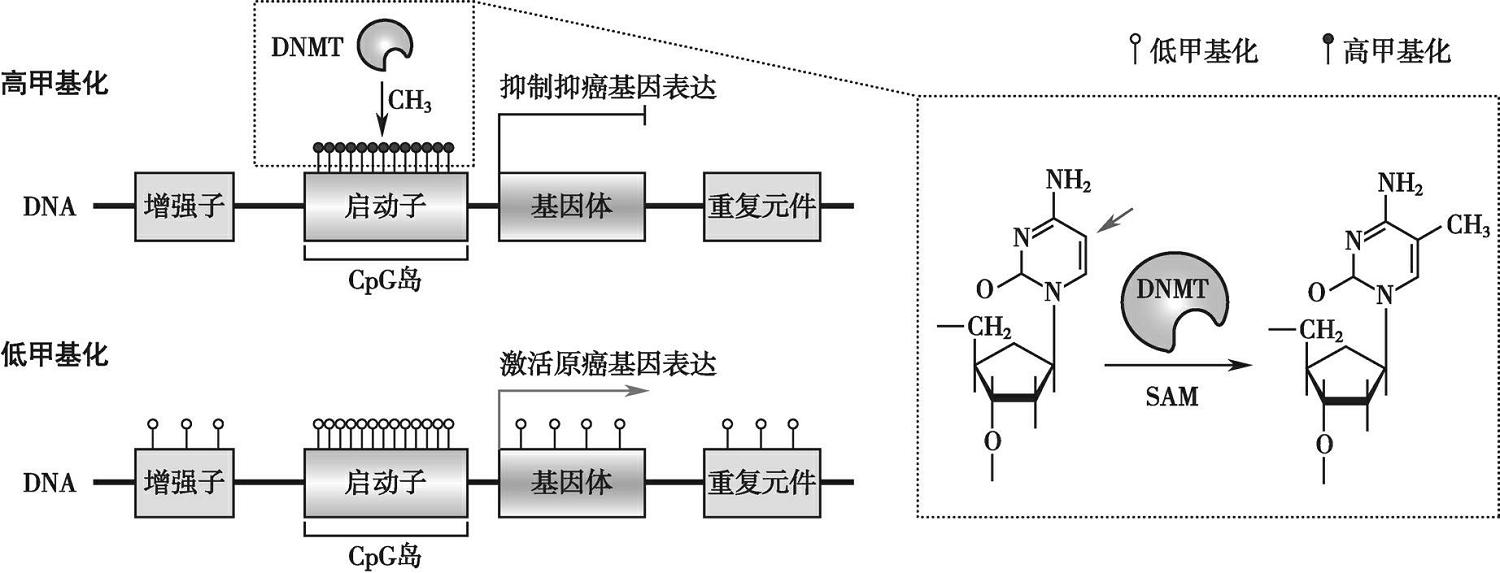
图3-7-1 DNA甲基化修饰的表观调控机制
启动子区CpG的高甲基化与癌细胞中的肿瘤抑制基因的转录抑制有关,在结直肠肿瘤中尤其明显,在一些重要的肿瘤抑制基因的启动子区域存在异常的高甲基化(表3-7-1)。
表3-7-1 一些CRC中发生高甲基化的基因
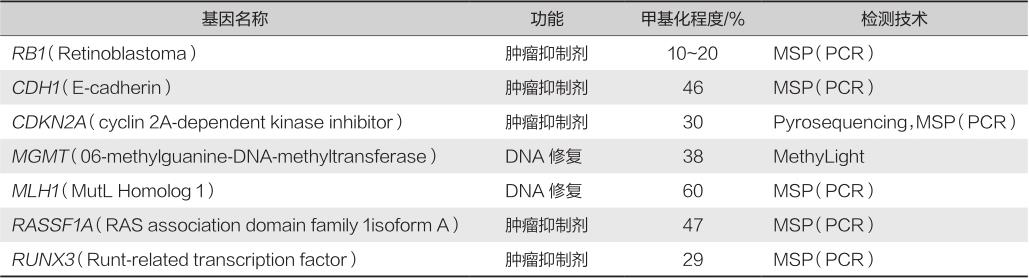
注:MSP.甲基化特异PCR;PCR.聚合酶链反应。
全基因组低甲基化是结直肠肿瘤中最早报道的异常甲基化事件之一,是结直肠肿瘤发生的早期事件。一般来说,三个区域的DNA低甲基化与结直肠肿瘤中原癌基因的激活有关:①启动子区域,该区域的低甲基化会导致基因印记的丢失或原癌基因的直接激活;②远距离调控区域,如增强子;③位于某些重复元件下游的反义启动子。人类基因组含有高达17%的含LINE 1型转座酶结构域1(LINE 1 type transposase domain containing 1)的重复元件,它们在正常生理条件下是沉默的,但如果通过低甲基化激活,便可通过“剪切和粘贴”机制发挥反转录转座子的作用,将自身插入远距离的脆弱位点(不稳定的基因组区域)并导致基因组不稳定。LINE-1的低甲基化与MSI和CIMP负相关,同时也与早发性结直肠肿瘤和预后不良有关,因此LINE-1成为潜在的结直肠肿瘤的重要生物标志物。
核小体是由核心组蛋白(histone,H)八聚体(H2A×2、H2B×2、H3×2、H4×2)与缠绕其外周的DNA组成的核心颗粒,以及颗粒之间DNA片段和一个H1构成。每个组蛋白的核心蛋白都具有富含赖氨酸和精氨酸残基的特征性尾部(N端),这些残基经过翻译后修饰,可以通过修饰的组蛋白-DNA相互作用直接影响基因表达,或通过改变特异性结合蛋白的识别位点间接影响基因表达。已有研究发现,组蛋白修饰与正常生理行为和发育过程中的许多细胞过程,以及包括癌症在内的各种疾病的发病机制有关,其中研究最多的主要是组蛋白乙酰化修饰和组蛋白甲基化修饰(图3-7-2)。
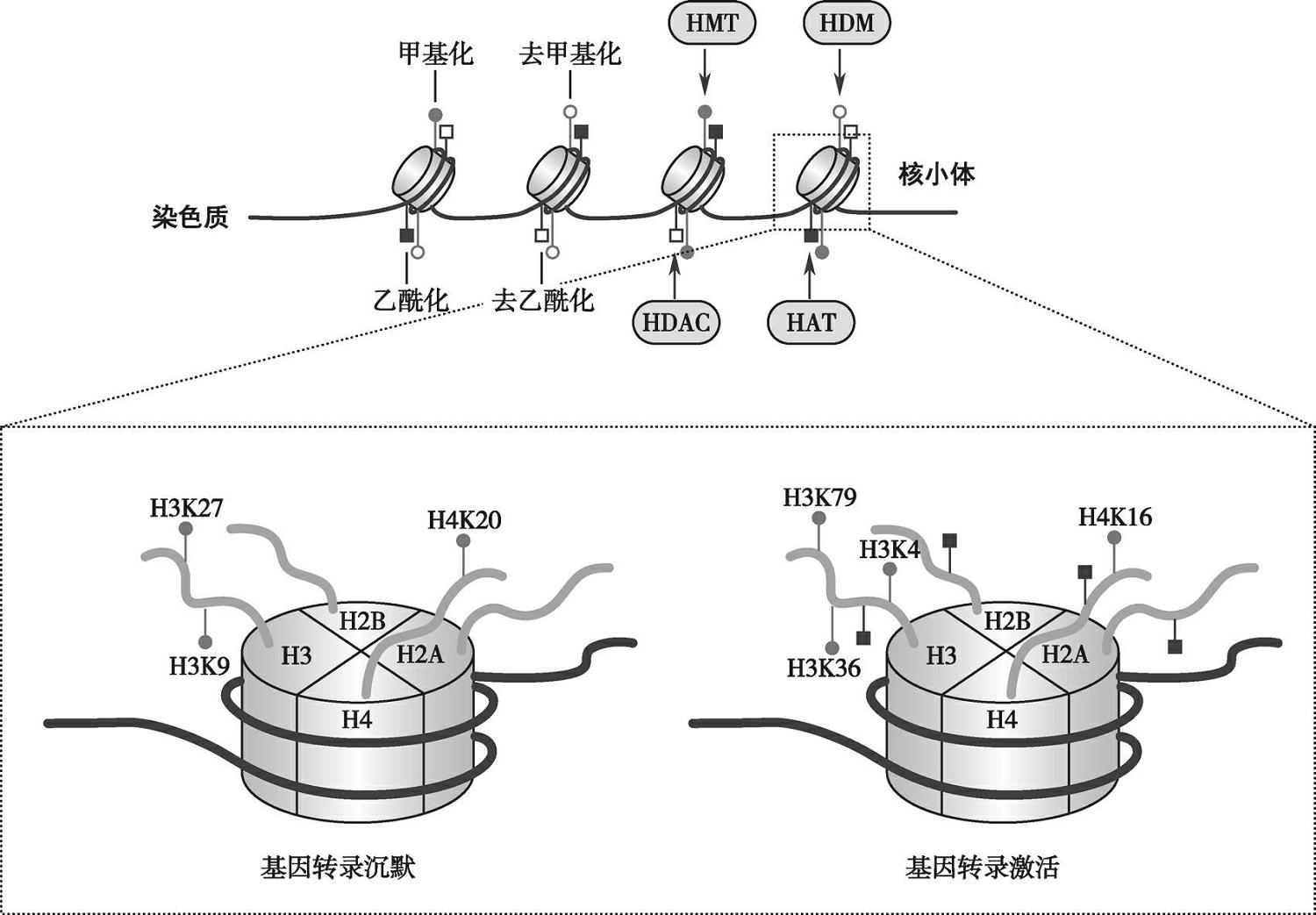
图3-7-2 组蛋白乙酰化和甲基化修饰的表观调控机制
组蛋白的乙酰化和去乙酰化分别由组蛋白乙酰转移酶和组蛋白脱乙酰酶催化。组蛋白乙酰转移酶催化乙酰基从乙酰辅酶A转移到组蛋白赖氨酸残基的氨基。组蛋白尾部的乙酰化可以中和带正电荷的赖氨酸,削弱尾部和带负电荷的核小体DNA之间的静电相互作用,从而影响染色质的压缩状态,使DNA更加容易转录。与原癌基因相关的组蛋白高度乙酰化,常会激活原癌基因表达;而与肿瘤抑制基因相关的组蛋白的低乙酰化,可使基因沉默。因此,这些组蛋白乙酰化调控酶作为CRC等恶性肿瘤的潜在治疗靶点引起了广泛关注。
组蛋白甲基化是指甲基以特定的方式添加到组蛋白H3和H4尾部的赖氨酸和精氨酸残基上,并且可以导致基因表达的激活或抑制。组蛋白甲基化和去甲基化由组蛋白甲基转移酶和组蛋白去甲基化酶催化,其过表达或低表达可能会改变整体组蛋白甲基化状态,改变数百种癌基因或肿瘤抑制基因的表达,并最终改变,促进癌症发展或进展。例如,赖氨酸特异性脱甲基酶1是一种组蛋白去甲基化酶(histone demethylase,HDMT),可脱甲基化H3K4me2和H3K9,参与调控CRC细胞增殖和分化。与组蛋白乙酰化相比,组蛋白甲基化不仅可以改变DNA的压缩状态,而且在染色质中产生可以被各种蛋白质识别的对接位点,例如包含转录复合物的蛋白质(如转录起始因子TFIID亚基3,可以激活Wnt/β联蛋白靶基因)。
在正常生理发育和几乎所有疾病的发病机制中,约98%的非蛋白质编码基因组参与了基因表达的调节,这些转录本通常被称为非编码RNA(non-coding RNA,ncRNA),可以在转录后进行剪接,但不能翻译成蛋白质。ncRNA能以组织特异性的方式发挥促肿瘤或抗肿瘤功能,已有研究表明,miRNA与lncRNA在CRC发生过程中发挥重要作用。
miRNA通过与其靶mRNA的3′-非翻译区中的互补序列结合而发挥转录后抑制的作用,调控超过60%的蛋白质编码基因的翻译。由于miRNA通常位于基因组内的脆弱位点,它们的表达可能通过各种遗传改变而失调,包括点突变、缺失、扩增或易位。此外,DNA高甲基化和低甲基化也可以改变miRNA的表达。许多研究已经发现了CRC肿瘤组织和相邻的正常组织之间miRNA不同的表达水平。miRNA可以通过抑制肿瘤抑制基因的表达作为致癌miRNA(onco-miRNA)发挥作用,或通过抑制癌基因表达作为肿瘤抑制miRNA(ts-miRNA)发挥作用。
lncRNA通过多种机制发挥转录的正向或负向调节作用,包括:与基因启动子或增强子的相互作用;通过充当染色质修饰蛋白复合物的指导分子来修饰染色质通路;核结构的监管;通过与靶mRNA和调节蛋白复合物直接相互作用来调节mRNA稳定性;作为miRNA海绵,通过lncRNA序列内的多个特异性结合位点凝集miRNA。大多数已知的与CRC相关的lncRNA都高表达,起miRNA海绵的作用。此外,也发现有少数lncRNA可以作为肿瘤抑制因子发挥作用,例如,与肿瘤相邻的正常组织相比,生长抑制特异性基因5(growth arrest-specific transcripts 5, GAS5 )在人类CRC组织中低表达,并且低水平的 GAS5 与肿瘤大小、晚期和较差的总生存期成正相关。lncRNA在许多癌症相关通路中发挥作用,如Wnt、EGFR、TGF-β和P53信号通路,并且可以影响CRC发生、进展和转移的几乎所有病理生理过程。
目前结肠镜检查可以检测和移除癌前病变,因此被认为是目前CRC筛查的金标准。然而,结肠镜检查是侵入性的、昂贵的,患者的依从性较低,同时也有出血和穿孔等潜在的并发症。相比之下,粪便隐血试验和粪便免疫化学试验是欧洲和其他西方国家最常用的非侵入性筛查试验,但对于癌前病变来说,其灵敏度和特异度低于结肠镜检查。因此,开发新颖且强大的非侵入性检测方法,对于发现癌前病变和早期CRC非常重要。此外,目前用于CRC分期的肿瘤淋巴转移(TNM)分类系统不足以预测预后,迫切需要发现能够区分复发和死亡可能性高的患者的预后生物标志物,以及可能真正受益于化疗、免疫治疗和/或靶向治疗的患者的预测性生物标志物。表观遗传标记及其调节因子,包括DNA甲基化、组蛋白修饰、miRNA和lncRNA,已显示出作为临床相关生物标记的前景,可用于CRC的诊断、预后和治疗反应预测。
DNA甲基化生物标志物用于诊断的效果明显,其中一些检测方法已商业化,用于当前的临床实践,甚至已进入临床指南。
血浆中 SEPT9 基因的甲基化是研究最为广泛的用于CRC诊断的DNA甲基化生物标志物,该基因的编码蛋白Septin9是一种参与肌动蛋白动态调控、细胞骨架重塑、囊泡运输和胞吐作用的GTP结合蛋白。多项研究分析了这种甲基化生物标志物在CRC大型队列中诊断的准确性,其灵敏度和特异度分别为48%~90%和73%~97%。血浆 SEPT9 基因的甲基化水平商业化为Epi proColo测试(Epigenomics),并于2016年被FDA批准为第一个基于分子血液的CRC筛查检测。然而,Epi proColon测试对于进展期(Ⅲ期~Ⅳ期)CRC患者的诊断准确度在统计学上显著优于早期(Ⅰ期~Ⅱ期)CRC患者。
另一种目前较为成熟的用于CRC诊断的非侵入性生物标志物是编码中间丝蛋白波形蛋白的 VIM 基因的甲基化,它与微管和肌动蛋白微丝一起构成细胞骨架。在血浆样本中, VIM 基因的甲基化的灵敏度和特异度分别高达59%和93%,在晚期疾病阶段灵敏度显著增加;在粪便样本中, VIM 基因的甲基化的灵敏度和特异度分别高达81%和95%。鉴于其在粪便样本中诊断准确性可能高于血液样本,该生物标志物也已商业化为ColoSure测试(LabCorp)。
组蛋白修饰作为生物标志物吸引力较低的原因包括:①其用作定量分析物相关的技术限制(因为大多数使用的方法,如免疫荧光或染色质免疫沉淀,不允许高通量分析);②对不同癌症缺乏特异性。相较于DNA甲基化,目前对组蛋白修饰作为生物标志物的研究较少,不过也有一些有诊断潜力的标志物。例如,与正常结肠黏膜相比,CRC和腺瘤中 H3K9 的甲基化水平显著升高,CRC中 H3K27 和 H4K12 的乙酰化修饰水平显著升高。对于非侵入性生物标记物的探索,有初步数据提示,与健康对照个体相比,在CRC患者中观察到循环核小体中 H3K9 三甲基化(H3K9me3)和 H4K20me3 标记水平降低。
miRNA作为候选生物标志物的潜力包括:①体积小、数量有限(相对于蛋白质编码基因);②在各种生物样本(如组织、血液和粪便)中较为稳定性;③常规实验室技术(如微阵列和定量逆转录PCR)几乎可以在所有标本类型中进行鉴定和量化。在过去10年中,研究CRC中miRNA的研究数量呈指数级增长,但只有少数研究是针对大型患者队列、精确定义的患者群体和独立验证队列进行的。尽管如此,已在CRC中鉴定出许多潜在的miRNA生物标志物。例如,miR-21的高表达可能是用于诊断、预后和预测CRC治疗反应的重要的生物标志物,其在血液和粪便中均表现出高灵敏度和特异度。与使用单个miRNA生物标志物相比,两个或多个miRNA组合可能是一种更好的诊断方法。
在过去的几年中,lncRNA作为CRC的生物标志物越来越受到关注。HOX转录反义RNA(HOX transcript antisense RNA,HOTAIR)在CRC发展的早期阶段,在血清和组织中均高表达,并且与TNM分期和患者生存相关。结直肠癌相关转录本1(colon cancer associated transcript 1,CCAT1)在肿瘤组织和血液中的高表达也是CRC发生的早期事件,并且与TNM分期、总生存期和无复发生存期相关。
对癌症表观遗传改变的研究不仅提供了有吸引力的生物标志物候选物,而且还为开发新的抗癌药物(即所谓的表观遗传修饰剂)打开了大门。与不可逆的遗传改变不同,从治疗的角度来看具有挑战性,表观遗传改变基本上是可逆的,使其成为有吸引力的治疗目标。已经开发了越来越多的表观遗传修饰剂,其中一些已获得FDA批准用于治疗各种疾病,但尚未有针对CRC的表观遗传修饰药物。已有的表观遗传修饰药物包括参与DNA甲基化(如DNMT和组蛋白脱乙酰酶)和组蛋白修饰(如去甲基化由组蛋白甲基转移酶和组蛋白去甲基化酶)的酶抑制剂,以及调节miRNA表达的治疗性药物,其中一些已经进入CRC的临床前试验或早期临床试验。
值得注意的是,如果单独使用和在晚期使用表观遗传修饰剂,可能导致表观遗传修饰剂的功效低下。在癌症的早期阶段,基因组改变的负担较低,并且表观遗传改变表现为癌变过程中的早期事件;因此,将表观遗传修饰剂用作癌症早期阶段的辅助治疗可能更有效,同时也可能需要延长治疗时间,因为细胞的重编程需要时间并且在治疗停止后可能不稳定。
近年来的科学研究表明,表观遗传学在肿瘤的发生和发展中发挥重要的调控作用。表观遗传学的改变可以作为驱动CRC发生的因素之一,也可以作为参与者调节正常结肠黏膜发展为CRC的恶性进程。与基因遗传突变不同,表观遗传学的改变具有动态性和可逆性,为CRC的预防、诊断、治疗及预后提供广阔的思路。更好地理解表观遗传调控机制,特别是肿瘤特异性的表观遗传改变,将有助于探索它们作为生物标志物的未来临床应用或它们作为CRC治疗靶点的潜力。
(谢珊珊)
[1]FEARON E R,VOGELSTEIN B.A genetic model for colorectal tumorigenesis[J]. Cell,1990,61(5):759-767.
[2]MAUGHAN T S,ADAMS R A,SMITH C G,et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer:results of the randomised phase 3 MRC COIN trial[J].Lancet,2011,377(9783):2103-2114.
[3]SOUGLAKOS J,PHILIPS J,WANG R,et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer[J].Br J Cancer,2009,101(3):465-472.
[4]RICHMAN S D,SEYMOUR M T,CHAMBERS P,et al.KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan:results from the MRC FOCUS trial[J]. J Clin Oncol,2009,27(35):5931-5937.
[5]REICHMANN A,MARTIN P,LEVIN B. Chromosomal banding patterns in human large bowel cancer[J]. Int J Cancer,1981,28(4):431-440.
[6]PELKA K,HOFREE M,CHEN J H,et al. Spatially organized multicellular immune hubs in human colorectal cancer[J].Cell,2021,184(18):4734-4752.
[7]LE D T,URAM J N,WANG H,et al. PD-1 blockade in tumors with mismatch-repair deficiency[J]. N Engl J Med,2015,372(26):2509-2520.
[8]WANG L,CUNNINGHAM J M,WINTERS J L,et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair[J]. Cancer Res,2003,63(17):5209-5212.
[9]TAKAYAMA T,KATSUKI S,TAKAHASHI Y,et al. Aberrant crypt foci of the colon as precursors of adenoma and cancer[J]. N Engl J Med,1998,339(18):1277-1284.
[10]KERR J F,WYLLIE A H,CURRIE A R. Apoptosis:a basic biological phenomenon with wide-ranging implications in tissue kinetics[J]. Br J Cancer,1972,26(4):239-257.
[11]ZHANG Z,ZHANG Y,XIA S,et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity[J].Nature,2020,579(7799):415-420.
[12]ASFAHA S,HAYAKAWA Y,MULEY A,et al. Krt19(+)/Lgr5(-)cells are radioresistant cancer-initiating stem cells in the colon and intestine[J]. Cell Stem Cell,2015,16(6):627-638.
[13]DE SOUSA E M F,DE SAUVAGE F J. Cellular plasticity in intestinal homeostasis and disease[J]. Cell Stem Cell,2019,24(1):54-64.
[14]MORRAL C,STANISAVLJEVIC J,HERNANDOMOMBLONA X,et al. Zonation of ribosomal dna transcription defines a stem cell hierarchy in colorectal cancer[J]. Cell Stem Cell,2020,26(6):845-861.
[15]ZHANG L,LI J,TIAN D,et al. Theranostic combinatorial drug-loaded coated cubosomes for enhanced targeting and efficacy against cancer cells[J]. Cell Death Dis,2020,11(1):1.
[16]LI S,PENG Y,PANCHENKO A R. DNA methylation:Precise modulation of chromatin structure and dynamics[J].Curr Opin Struct Biol,2022,75:102430.
[17]AUDIA J E,CAMPBELL R M. Histone modifications and cancer[J]. Cold Spring Harbor perspectives in biology,2016,8(4):a019521.
[18]GARGALIONIS A N,PIPERI C,ADAMOPOULOS C,et al. Histone modifications as a pathogenic mechanism of colorectal tumorigenesis[J]. Int J Biochem Cell B,2012,44(8):1276-1289.