
下载掌阅APP,畅读海量书库
立即打开



关于双原子分子的原子间的力能够作出的最简单的假定是:力正比于核间距r和它的平衡值r e 的差值. 因为U在r e 处有一极小,从而在r e 附近,
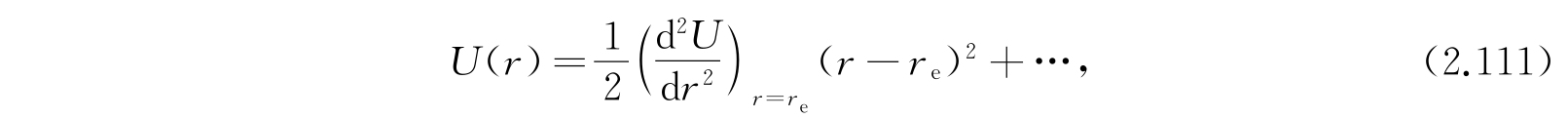
在r e 处U取作零,所以能量W是从U(r e )量起的. 令
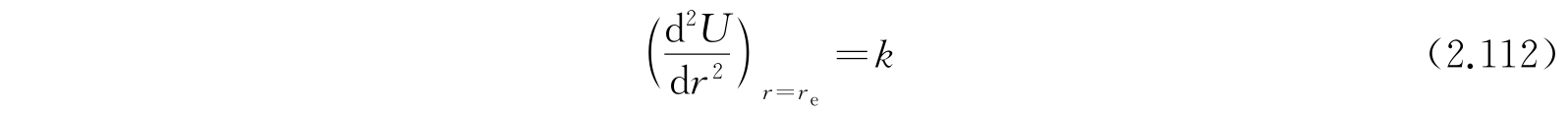
和
ρ=r-r e , (2.113)
由方程(2.110)得到
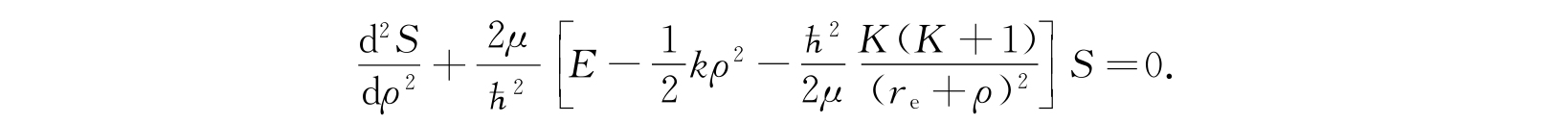
由于这一近似只对小ρ值适用,因此上述方程中最后一项应该展成
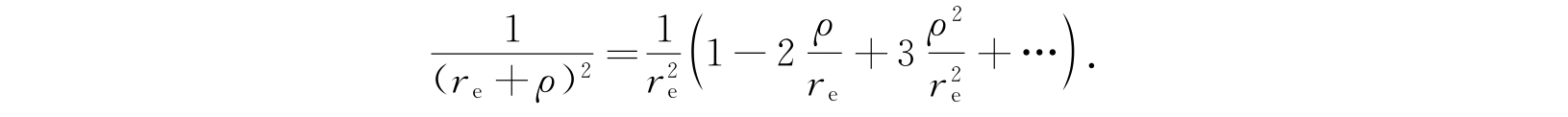
保留到ρ的平方项,方程可以写作
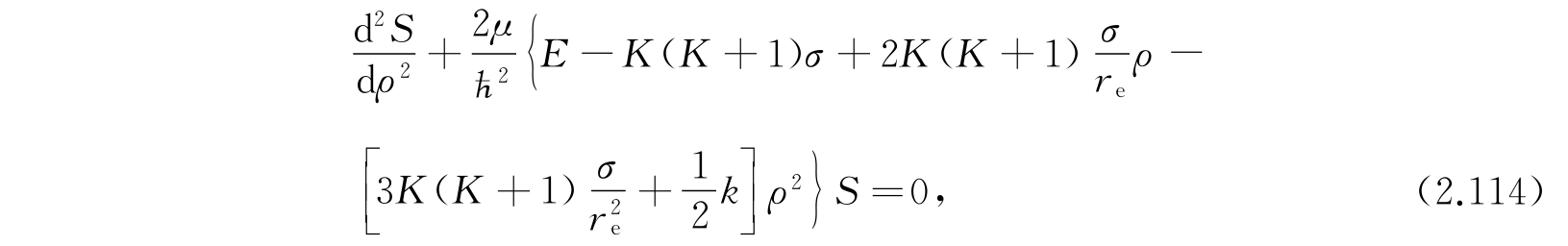
其中
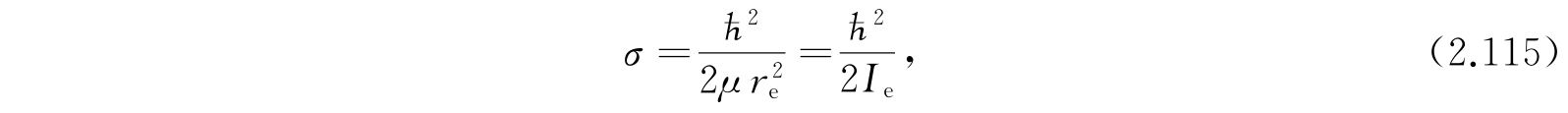
I e 是分子转动惯量的平衡值.
利用
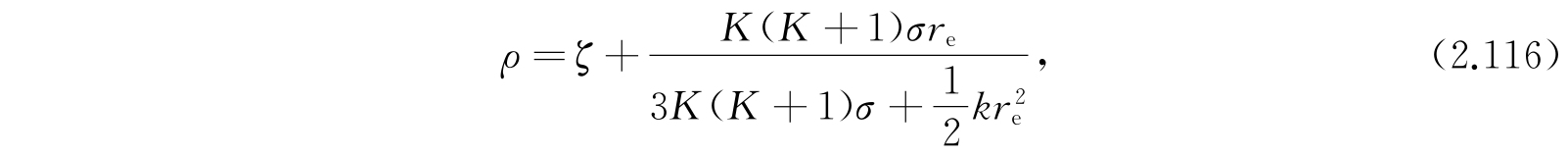
我们能够把式(2.114)化为式(2.25)那样的谐振子方程:
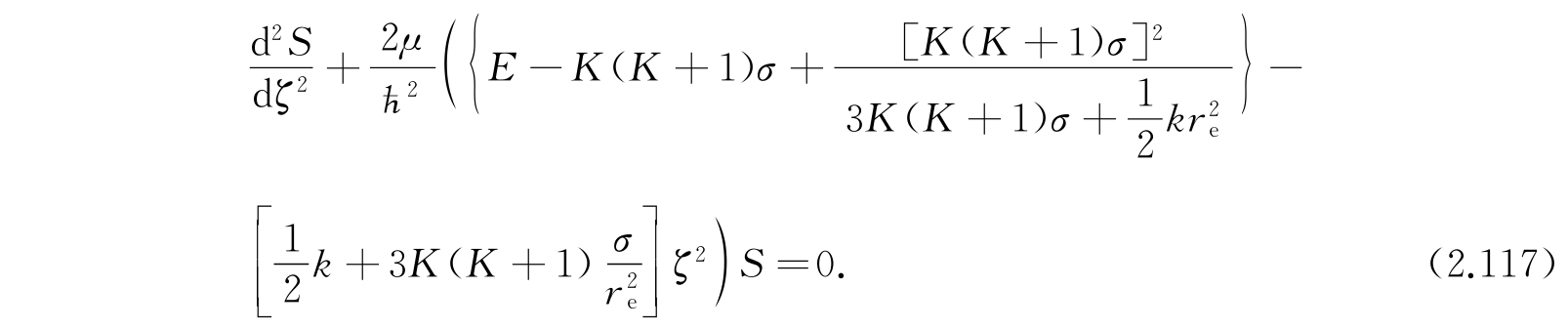
边界条件是在r=0和r=∞时S等于零. 这一条件和谐振子的边界条件有些不同:对谐振子在ζ=±∞时S是零. 但是因为S在经典许可的区域以外迅速地减少,把这两个条件看作等价,实际上也并不会引进严重的误差,所以我们可以用谐振子的波函数作为函数S的近似.
于是能级为
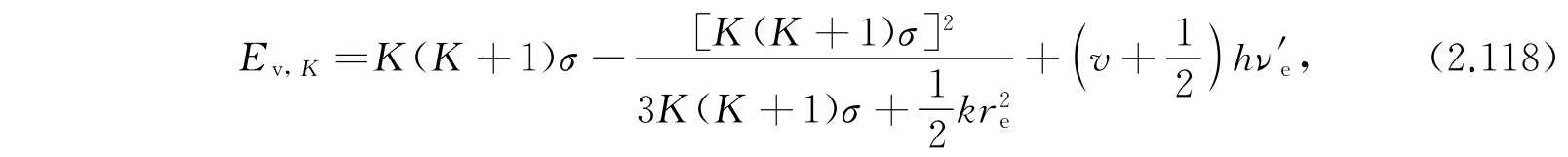
其中
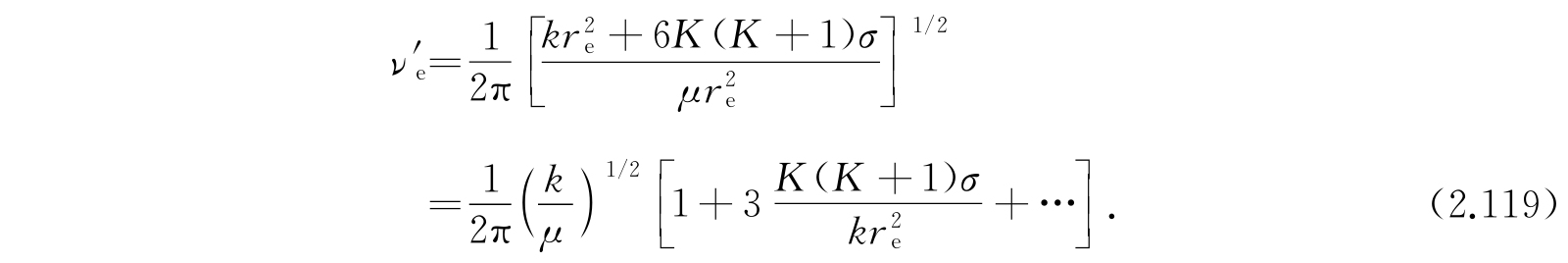
如果我们引入在平衡状态附近的振动频率ν e ,
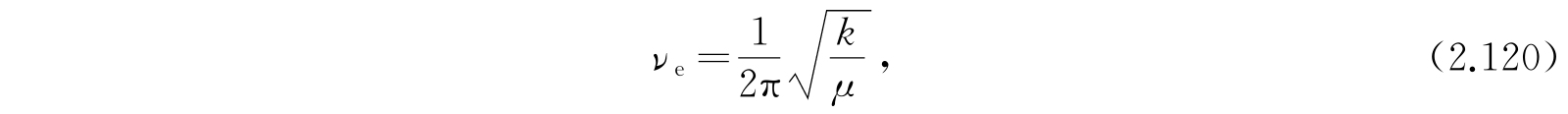
就得到最终的近似公式
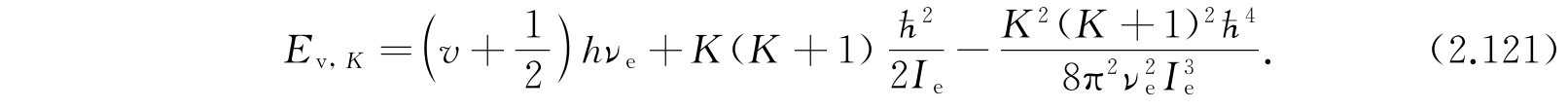
第一项显然是分子被看作谐振子的振动能;第二项是假定分子为刚体时的转动能;第三项的来源是:考虑非刚性分子由于转动产生分子的伸长,伸长后角惯量又有所改变,因此又产生这个修正项. 更高次的项并不可靠,因为所假定的势函数不够精确. 大多数分子的实验数据很好地适合方程(2.121). 对于更精确的工作,还需要其他的项并且需要比式(2.111)更精确的势函数.
前面已经说过,量子数K是和分子的转动关联的. 但是现在得到的转动能是
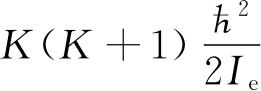 . 若p
θ
是转动的角动量,就正如线性动量的情形,转动能是
. 若p
θ
是转动的角动量,就正如线性动量的情形,转动能是
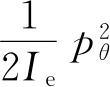 . 所以对任何特殊K值,角动量是
. 所以对任何特殊K值,角动量是
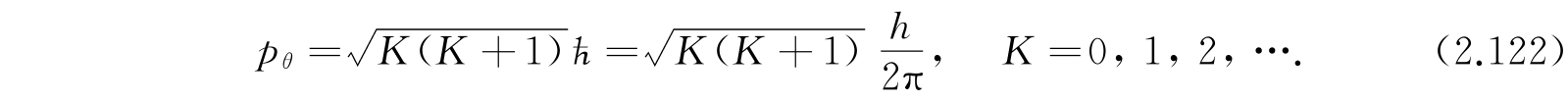
因为按照方程(2.107),有2K+1个M值与各个不同的波函数对应,也与不同的转动轴定向对应,所以转动态是2K+1重退化的. 事实上发现在任意特别选定的方向,例如分子的轴(取作z轴)上角动量的分量是
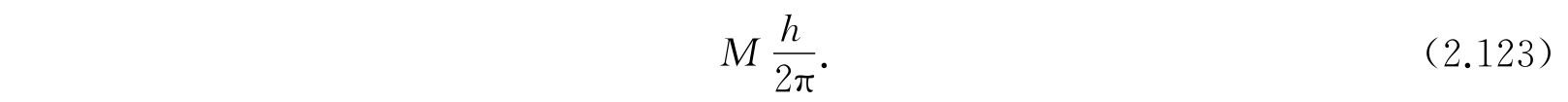
这些结果对任何具有转动惯量为I e 的刚性线转子都是对的. 需要着重指出的是:按照方程(2.115),决定I e 的是平衡核间距和核质量,而不是原子的质量.
前一节所给出的简单处理与实验有一些不符合的地方,上节给出的是有均匀间隔的能级,而实验观察到的振动能级却显示出随v增加而收敛. 为了获得这样一个特点,就需要用比前节中的势函数更接近于真实的势函数U(r),特别要注意对大r值时势函数的行为.
莫尔斯(P.M. Morse)建议了下列形式的势函数:
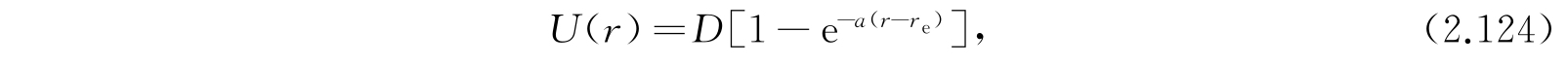
它在r=r e 处有极小值为零,对大的r值趋近于某一有限值D. 在r=0处,实际的势能函数应该是无穷大,但莫尔斯势却是有限的. 不过,莫尔斯势在这一点的值也很大,所以这一缺陷并不严重.
将式(2.124)代入式(2.110),我们得到
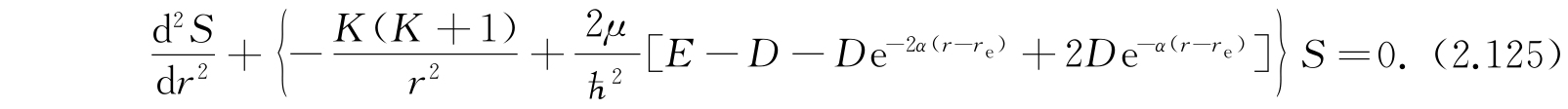
我们作下列替换:
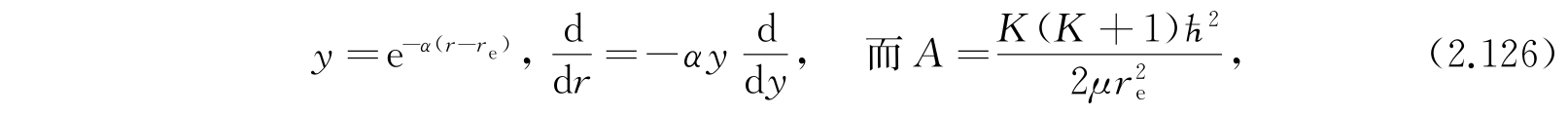
方程变为
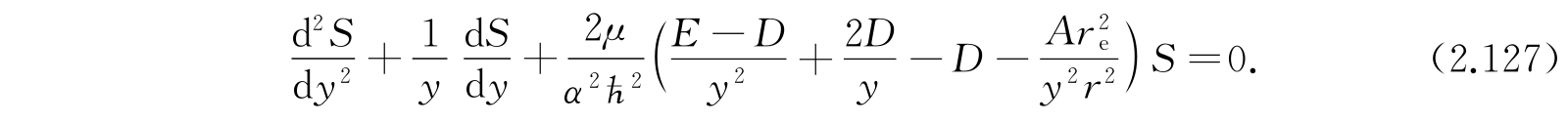
最后一项是转动和振动型的耦合效应,所以在低转动和振动态时是很小的. 在平衡时r=r e ,y=1,我们把(r e /r) 2 展成(y-1)的幂级数:
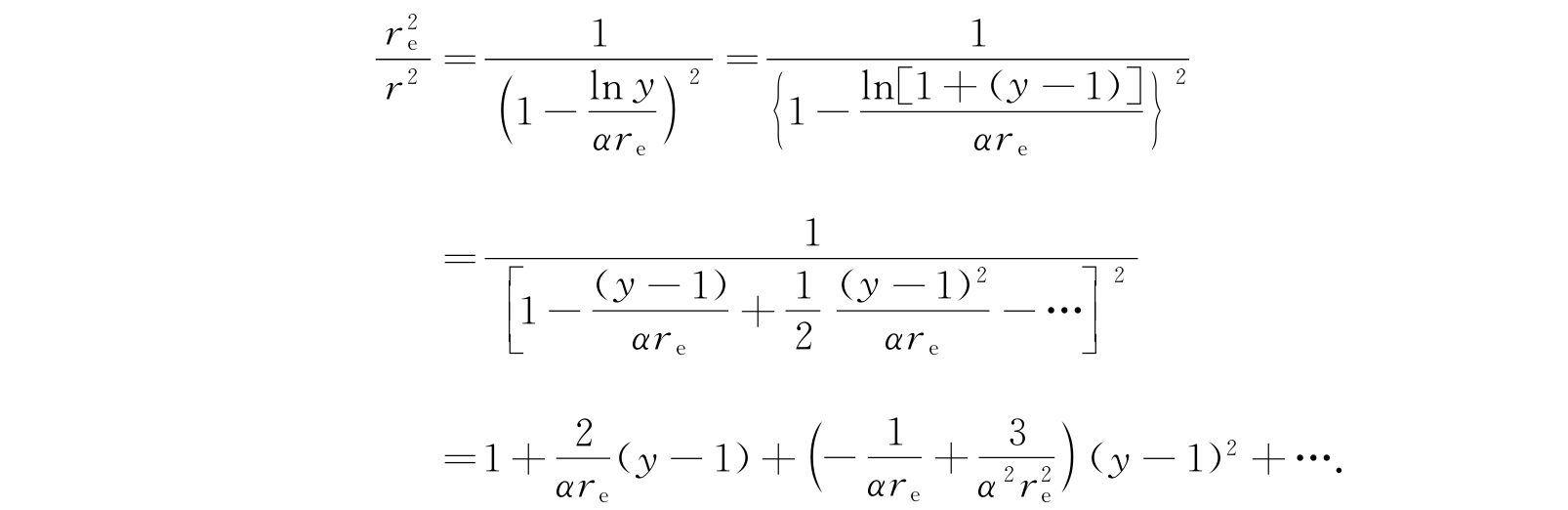
只用已经写出的前三项,方程(2.127)就变成
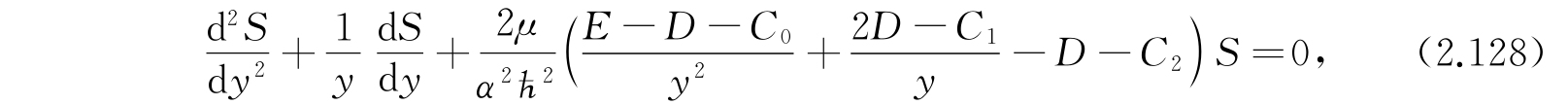
其中
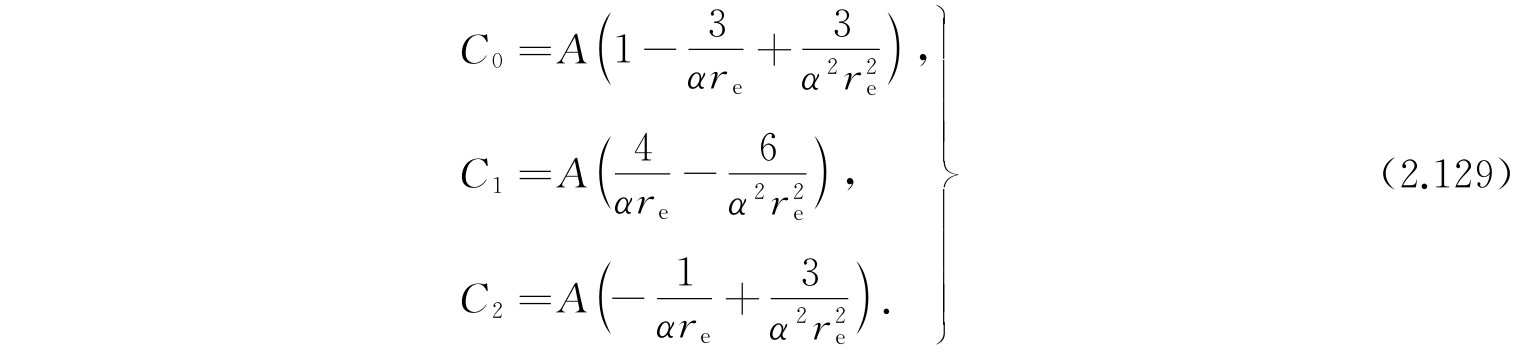
现在用下列替换可以把式(2.128)化成前面处理过的氢原子的径向函数方程:
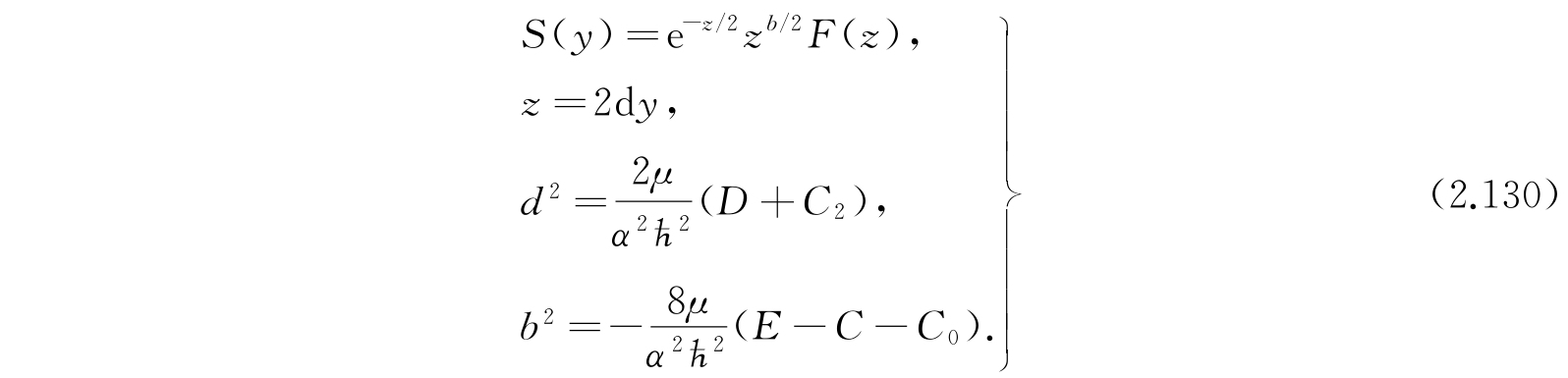
于是就有
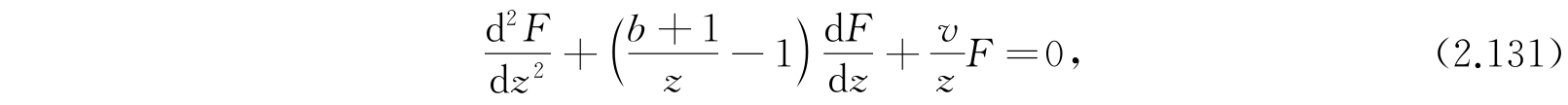
其中
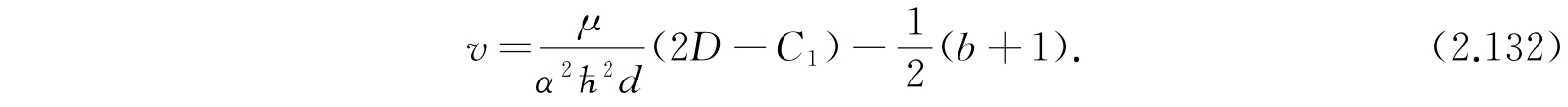
这一方程与氢原子的半径波函数的方程很相似,并且能够用拉盖尔多项式解.v的本征值是0,1,2,…. 利用式(2.130)和(2.132)解E,得
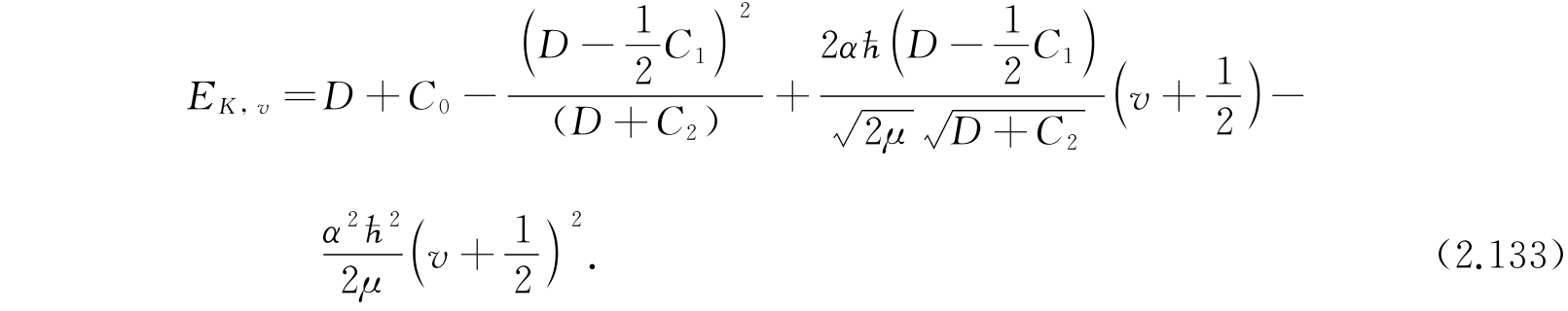
因为A是很小的,因此我们应该把上面的表示式展成C 1 /D和C 2 /D的幂次项,于是
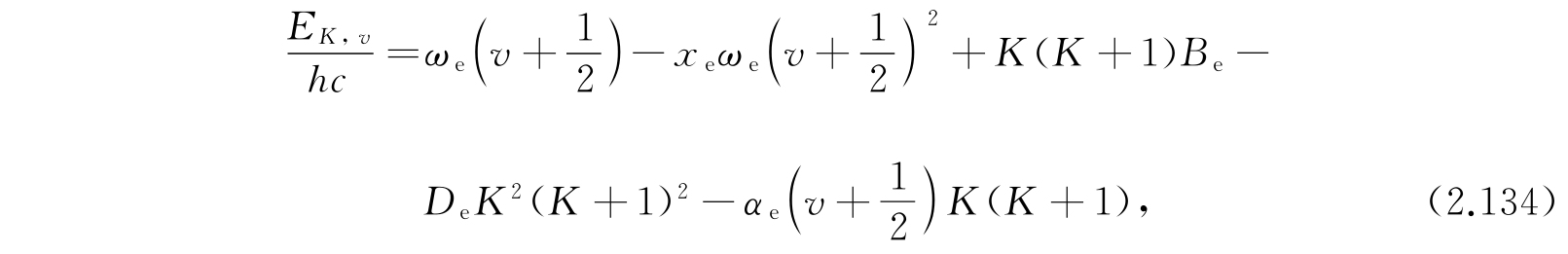
而
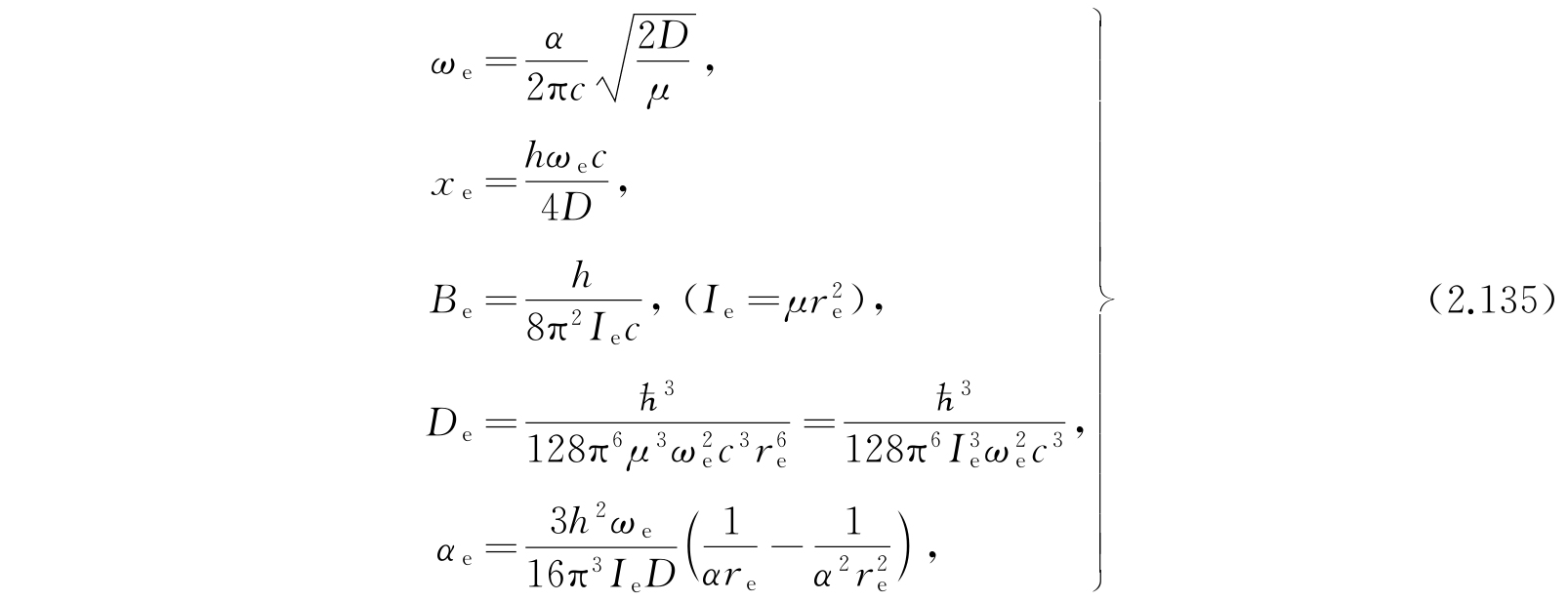
其中c是真空中的光速.
这一关系式几乎对所有双原子分子都能给出非常精确的能级数值. 无须说明,所有的计算是关于较低电子能级的. 式(2.134)中的常数的实验值曾经由赫兹堡(G. Herzberg)
 给出. 根据假定的势函数V(r),显然由式(2.134)计算的E只当E<D时才有意义. 对E>D,按照2.2节的一般讨论,能级应该是连续的而不是间断的. 所以间断能级的数目是有限的. 在应用式(2.134)时必须记住这一事实. 基态(v=0,K=0)的能量是
给出. 根据假定的势函数V(r),显然由式(2.134)计算的E只当E<D时才有意义. 对E>D,按照2.2节的一般讨论,能级应该是连续的而不是间断的. 所以间断能级的数目是有限的. 在应用式(2.134)时必须记住这一事实. 基态(v=0,K=0)的能量是
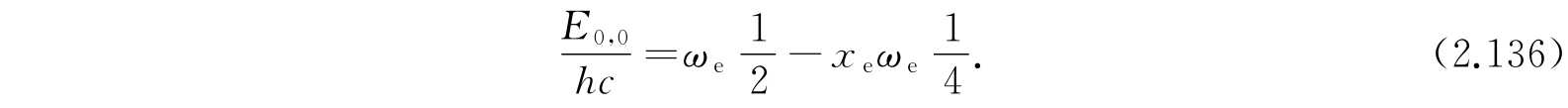
E K,v 和E 0,0 的差值是
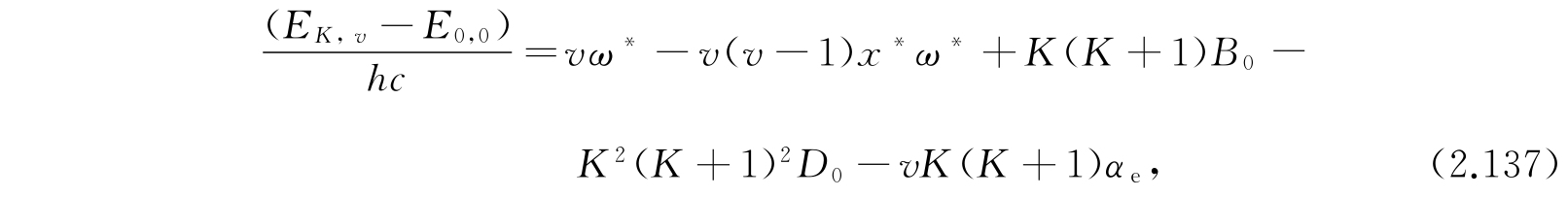
其中
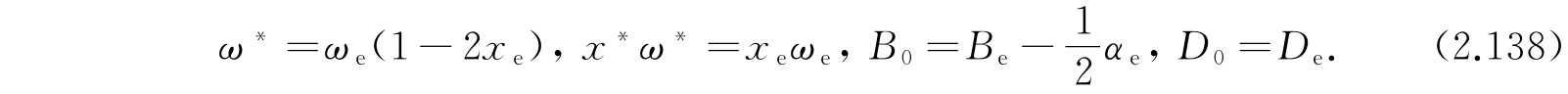
离解能是D-E 0,0 ,即
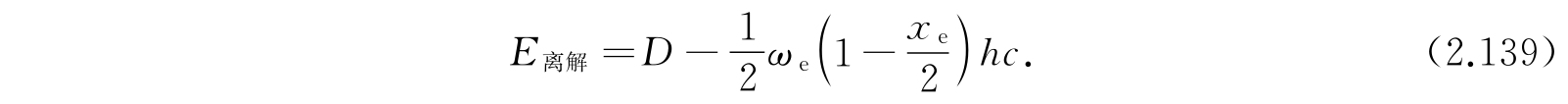
D可以按照式(2.135)由ω e 和x e 计算出来,所以离解能也可以从光谱数据计算出来. 但是式(2.135)由各种近似得到,这样测定的离解能不可能希望是准确的;实际上也常和用更直接的方法所确定的数值不很一致. 我们可以举一个例子,考虑氯化氢(HCl)分子,就有

实验值是102.1 kcal/mol(1 cal=4.186 8 J),约有17%的误差,但作为估计则是够用的了. 这说明从光谱的数据,我们能估计分子的离解能.