
下载掌阅APP,畅读海量书库
立即打开



DNA聚合酶的校对功能可保证DNA复制的保真性,对遗传信息在细胞增殖时的准确传递至关重要。DNA复制虽然极少出错但并非万无一失。此外,即使在非复制期间,DNA也会发生损伤,导致DNA的序列或结构出现异常,甚至发生基因突变。这种突变会影响表型,一方面是生物进化的基础,另一方面又是个体患病甚至死亡的遗传因素。不过,在漫长的进化过程中,生物体已经建立了一组修复系统,可以修复各种DNA损伤,以保证生命的延续性和遗传的稳定性。
DNA复制的保真性使生物体维持着遗传信息的稳定性。不过,稳定是相对的,变异是绝对的。变异即 基因突变 (mutation),包括 静态突变 (static mutation,包括点突变和片段突变,能稳定遗传给子代)和 动态突变 (dynamic mutation,主要是重复序列拷贝数增加,遗传给子代时进一步发生突变)。基因突变的化学本质是 DNA损伤 (DNA damage),是指DNA结构出现异常,导致细胞或病毒的基因型发生稳定的、可遗传的变化,这种变化有时导致基因产物功能的改变或缺失,从而导致细胞转化或死亡。
DNA损伤会导致基因突变,一方面有利于生物进化,另一方面又可能产生不利后果。
1.突变是生物进化的分子基础 遗传与变异是对立统一的生命现象。突变容易被片面理解成会危害生命,但实际上突变的发生在各种生物体内普遍存在,并且有其积极意义。有突变才有生物进化,没有突变就不会有大千世界的生物多样性。
2.致死突变消灭有害个体 致死突变 (lethal mutation)发生在对生命过程至关重要的基因上,可导致细胞死亡或个体夭亡,消灭病原体。例如短指是一种隐性致死突变,其纯合子个体会因骨骼缺陷而夭亡。
3.突变是许多疾病的分子基础 在致病突变中,点突变占70%(其中错义突变占49%,无义突变占11%,剪接位点突变占9%,调控元件突变占1%),插入缺失突变占23%,重排等占7%。
肿瘤细胞突变远多于其他细胞,其中赋予肿瘤细胞生长优势,因而与细胞癌变相关的不到0.1%,称为 驱动突变 (driver mutation),其余超过99.9%部分称为 乘客突变 (passenger mutation)。驱动突变涉及约65种癌基因、75种抑癌基因,包括影响细胞生存的(如 ras 、 PIK3CA 、 MAPK ),影响基因组稳定性的(如 ATM 、 ATR ),影响细胞归宿,即分裂、分化或静息的(如 APC )。仅有少数突变基因在多种肿瘤中都常见,如 ras 、 p53 、 RB1 (第九章,243页)。
4.突变是多态性的分子基础 例如单核苷酸多态性。
DNA损伤类型多种多样,其中有些损伤可以遗传,因此导致基因突变。
1.错配(mismatch) 导致DNA链上的一个碱基对被另一个碱基对置换,称为 碱基置换 (base substitution)(图2-28)。碱基置换有两种类型:① 转换 (transition),是嘧啶碱基之间或嘌呤碱基之间的置换,这种方式占2/3,其中又以C→T转换最多,发生率约为其他转换的10倍。② 颠换 (transversion),是嘌呤碱基和嘧啶碱基之间的置换。

图2-28 错配和插入缺失
2.插入缺失(indel) 是指DNA序列中发生一个碱基对或一小段核苷酸序列(通常是1~60bp)的插入缺失。插入缺失位点如果位于编码区内(第四章,99页),且插入缺失的不是3 n 个碱基对,会导致该位点下游的遗传密码全部发生改变,这种突变称为 移码突变 (frameshift mutation,图2-28);插入缺失的如果是3 n 个碱基对,则突变位点下游的遗传密码不会改变,这种突变称为 整码突变 (in-frame mutation)。
由一个碱基对的置换或插入缺失所导致的突变统称 点突变 (point mutation)。如果编码区发生点突变,会导致遗传密码改变,有多种可能的结果。
(1) 错义突变 (missense mutation):是指一种氨基酸的密码子突变成为另一种氨基酸的密码子,约占第三碱基置换的25%。
镰状细胞贫血 是点突变致病的典型例子:患者镰状血红蛋白β亚基基因的编码序列有一个点突变A→T(腺嘌呤被胸腺嘧啶置换),使原来6号谷氨酸(记作Glu6)密码子GAG(或GAA)变成缬氨酸密码子GTG(或GTA)(记作Glu6Val)。
(2) 无义突变 (nonsense mutation):又称终止突变(stop mutation),是指一种氨基酸的密码子突变成为终止密码子,导致翻译提前终止,所编码的蛋白质通常完全失活。突变成为终止密码子UAA、UAG、UGA的无义突变分别称为赭石突变、琥珀突变、乳白突变。
(3) 同义突变 (synonymous mutation):又称中性突变(neutral mutation),是指氨基酸的密码子突变成为其另一种同义密码子,因此不影响蛋白质结构,属于 沉默突变 (silent mutation,也有学者定义同义突变与沉默突变同义),占编码区错配的50%。
(4) 移码突变 (frameshift mutation):插入缺失一个碱基对导致的移码突变属于点突变。
3.重排(rearrangement) 又称基因重排、DNA重排、 染色体易位 (chromosomal translocation),是指基因组中较大DNA片段(10~1000bp)移动位置,但不包括基因组DNA缺失或外源DNA插入。重排可发生在DNA分子内部(染色体内),也可发生在DNA分子之间(染色体间),例如血红蛋白Lepore病就是重排的结果(图2-29)。
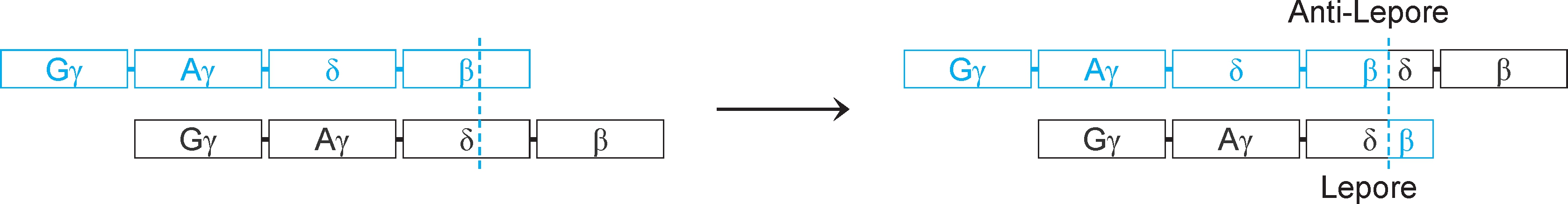
图2-29 重排与血红蛋白Lepore病
● 中期染色体存在一种 脆性位点 (fragile site),该位点的DNA容易断裂、缺失或重排。有的脆性位点含短串联重复序列。例如, 脆性X综合征 (fragile X syndrome)是一种X 连锁显性遗传病,表现为中度至重度智力低下,面部特征异常(如长脸、大耳、突颚等)。患者 X染色体上的一种翻译抑制因子基因 FMR1 (脆性X智力低下基因1)的5'非翻译区(第四章,99页)存在含CGG短串联重复序列的脆性位点,所含CGG短串联重复序列拷贝数高达200~3000个,而正常人只有6~50个,女性携带者和男性传递者为50~200个。
4.共价交联 是指碱基之间形成共价键连接。例如,①同一股DNA链上相邻的胸腺嘧啶发生共价交联(链内交联),形成胸腺嘧啶二聚体,会抑制复制和转录。②补骨脂素类(psoralen)可在双链之间形成交联(链间交联),会抑制复制和转录。
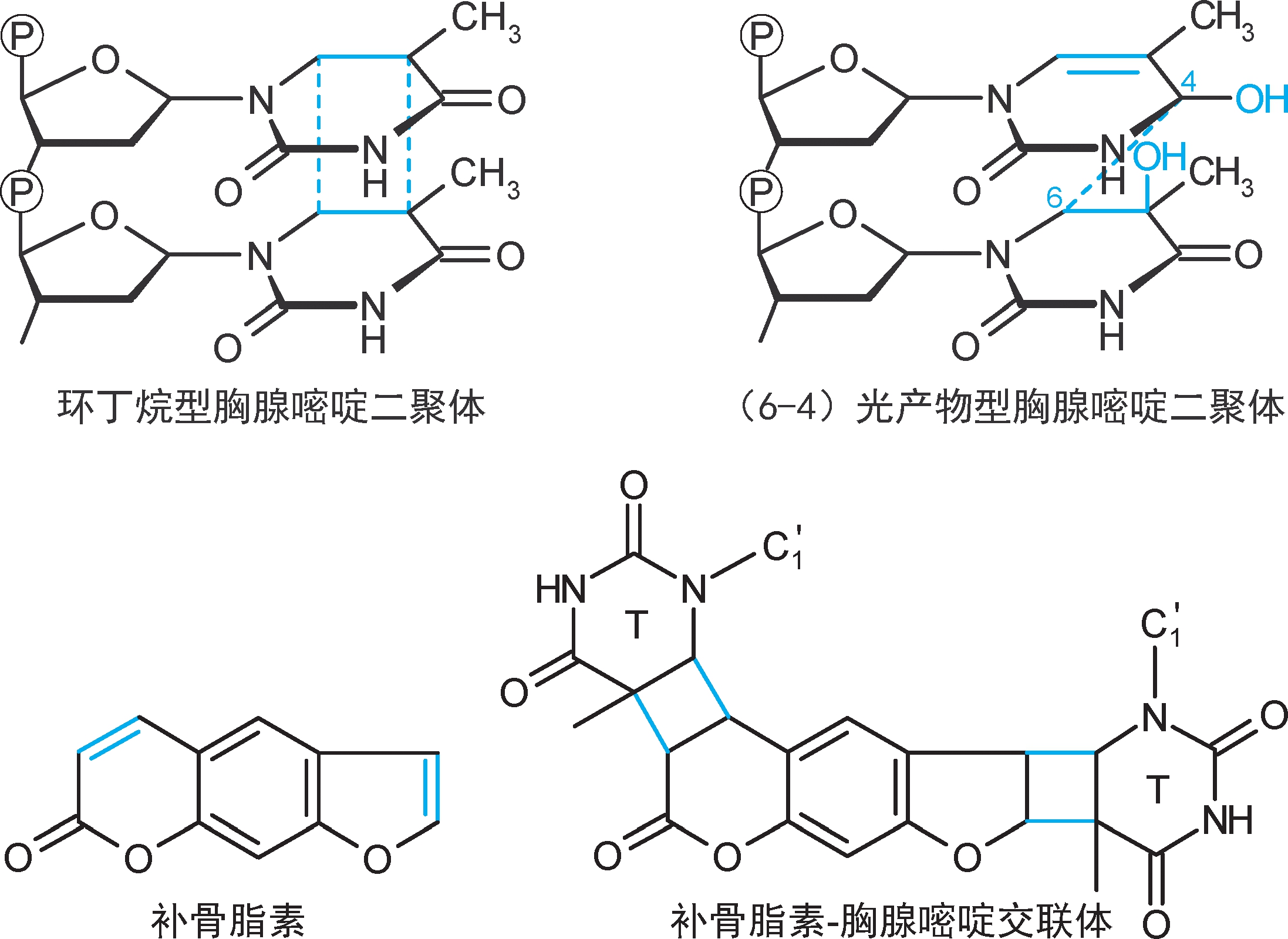
5.单碱基损伤 ①脱碱基:糖苷键非酶促水解脱去碱基(脱嘌呤多于脱嘧啶),形成无碱基位点(abasic site,apurinic or apyrimidinic site, 无嘌呤嘧啶位点 ,AP site, AP位点 )。其中脱嘌呤日发生率约为1/10 5 ,一个哺乳动物细胞DNA每日可脱5000~10000个。②脱氨基:胞嘧啶非酶促脱氨基成尿嘧啶(尿嘧啶被糖苷酶脱去),日发生率约为1/10 7 ,一个哺乳动物细胞DNA每日可脱100~500个;腺嘌呤脱氨基成次黄嘌呤(次黄嘌呤与C形成碱基配对);鸟嘌呤脱氨基成黄嘌呤。③碱基烷化:黄曲霉毒素将鸟嘌呤烷化为7-加成物,诱导G→T颠换。④氧化:羟自由基将鸟嘌呤氧化为8-氧鸟嘌呤(8-oxoG),后者在DNA合成时会与腺嘌呤配对,诱导G→T颠换。
6.主链断裂 电离辐射、自由基或某些化学试剂(如博莱霉素)可以使磷酸二酯键断裂,从而导致DNA单链断裂或双链断裂。
值得注意的是,绝大多数致病突变发生在编码区(其中60%为碱基置换,20%~25%为插入缺失),仅有不到1%发生在调控区(调控元件)。
内部因素和外部因素都会引起DNA损伤。内部因素如复制错误、自发性损伤会产生 自发突变 ,特点是突变率相对稳定。例如细菌的碱基对突变率10 -10 ~10 -9 /代,基因(1000bp)突变率约10 -6 /代,基因组突变率约3×10 -3 /代,高等生物基因组突变率10 -8 ~10 -5 /代。人类基因组突变率10 -7 ~10 -6 /代,外部因素如物理因素、化学因素、生物因素会引起 诱发突变 ,这些因素被称为诱变剂(致突变原,mutagen)。
1.复制错误 主要导致点突变。DNA聚合酶选择核苷酸的错配率为10 -5 ~10 -4 ,经过3'→5'外切酶活性校对降至10 -8 ~10 -6 。
DNA复制时,由于DNA聚合酶偶尔“打滑”(slippage, 复制滑动 、 复制滑移 ),模板或新生链会发生核苷酸的“环出”现象。新生链环出会导致子二代DNA发生插入,而模板环出会导致子二代DNA发生缺失。发生复制滑动的主要位点是重复序列,特别是短重复序列(图2-30)。例如,结肠癌TGF-β受体RⅡ常见一种突变,其基因序列中存在一段由10个连续的腺苷酸组成的短重复序列(A 10 )。在复制时,由于DNA聚合酶滑动,子代DNA中的该短重复序列长度会改变至A 9 或A 11 。

图2-30 复制滑动
2.自发性损伤 DNA分子可以由于各种原因发生化学变化。碱基发生酮-烯醇互变异构是导致自发突变的主要原因,此外还有碱基修饰、碱基脱氨基甚至碱基丢失等。这些变化会影响碱基对氢键的形成,因而影响碱基配对。如果这些变化发生在DNA复制过程中,就会发生错配。
3.物理因素 电离辐射(γ射线、X射线)和非电离辐射(紫外线及更长波长的电磁波)可导致主链断裂或交联、碱基丢失等。紫外线(特别是100~280nm的UVC)通常使DNA链上相邻的嘧啶碱基形成二聚体(人皮肤细胞每小时发生5×10 4 处,T 2 最多,C 2 最少),在局部扭曲DNA双螺旋结构,阻断复制和转录。电离辐射例如X射线可直接使DNA主链断裂,也可作用于水而生成活性氧(氧化应激时生成增加),间接导致DNA断链或碱基氧化。在外部因素引起的DNA损伤中有10%是由紫外线和电离辐射引起的。
4.化学因素 碱基类似物、碱基修饰剂(烷化剂、黄曲霉毒素)、染料、芳香族化合物、肿瘤化疗药物等可以引起DNA损伤。
(1)碱基类似物:氟尿嘧啶(5-FU,XD06)通过补救途径转化为嘧啶核苷酸类似物,在DNA合成时可代替正常核苷酸掺入DNA。这些类似物容易发生互变异构,引起错配。所有碱基类似物引起的错配均为转换。
(2)碱基修饰剂:通过修饰碱基改变碱基配对,例如羟胺、亚硝酸盐(诱导A→G转换、C→T转换)、烷化剂及活性氧(诱导G→T颠换)等自由基。
烷化剂是极强的诱变剂,它们带有一个或多个活性烷基,可以将DNA碱基烷基化,烷基化反应主要发生在鸟嘌呤的N-7位和腺嘌呤的N-3位上。①烷基化鸟嘌呤不稳定,容易水解脱落,从而形成AP位点,它会改变碱基配对性质,或者干扰DNA合成,例如导致缺失。②烷化剂还能使鸟嘌呤交联成二聚体,或者使DNA双链交联。交联DNA无法修复,因而烷化剂毒性较大,能导致细胞癌变、肿瘤发生。例如氮芥类(环磷酰胺、苯丁酸氮芥、苯丙氨酸氮芥)、硫芥、硫酸二甲酯、烷基磺酸盐、环氧化物类(环氧乙烷,苯并芘、黄曲霉毒素B 1 转化产物)、卤代烃(溴代甲烷)。
某些抗肿瘤药物(XL01)作用机制即致DNA损伤,如烷化剂类(XL01A)中的氮芥类似物(XL01AA,氮芥、环磷酰胺、苯丁酸氮芥)、烷基磺酸盐(XL01AB,白消安)、亚硝基脲类(XL01AD,司莫司汀)、其他烷化剂(XL01AX,达卡巴嗪)。
(3)嵌入剂:原黄素、吖啶黄、吖啶橙、溴化乙锭等化合物有扁平芳香环结构,可以嵌入双链DNA相邻碱基对之间,所以称为 嵌入染料 (intercalative dye)。它们与碱基对大小相当,嵌入之后会引起复制滑动,发生插入缺失,从而导致移码突变。
5.生物因素 病毒DNA整合、转座子转座(68页)等可以改变基因结构,或者改变基因表达活性。
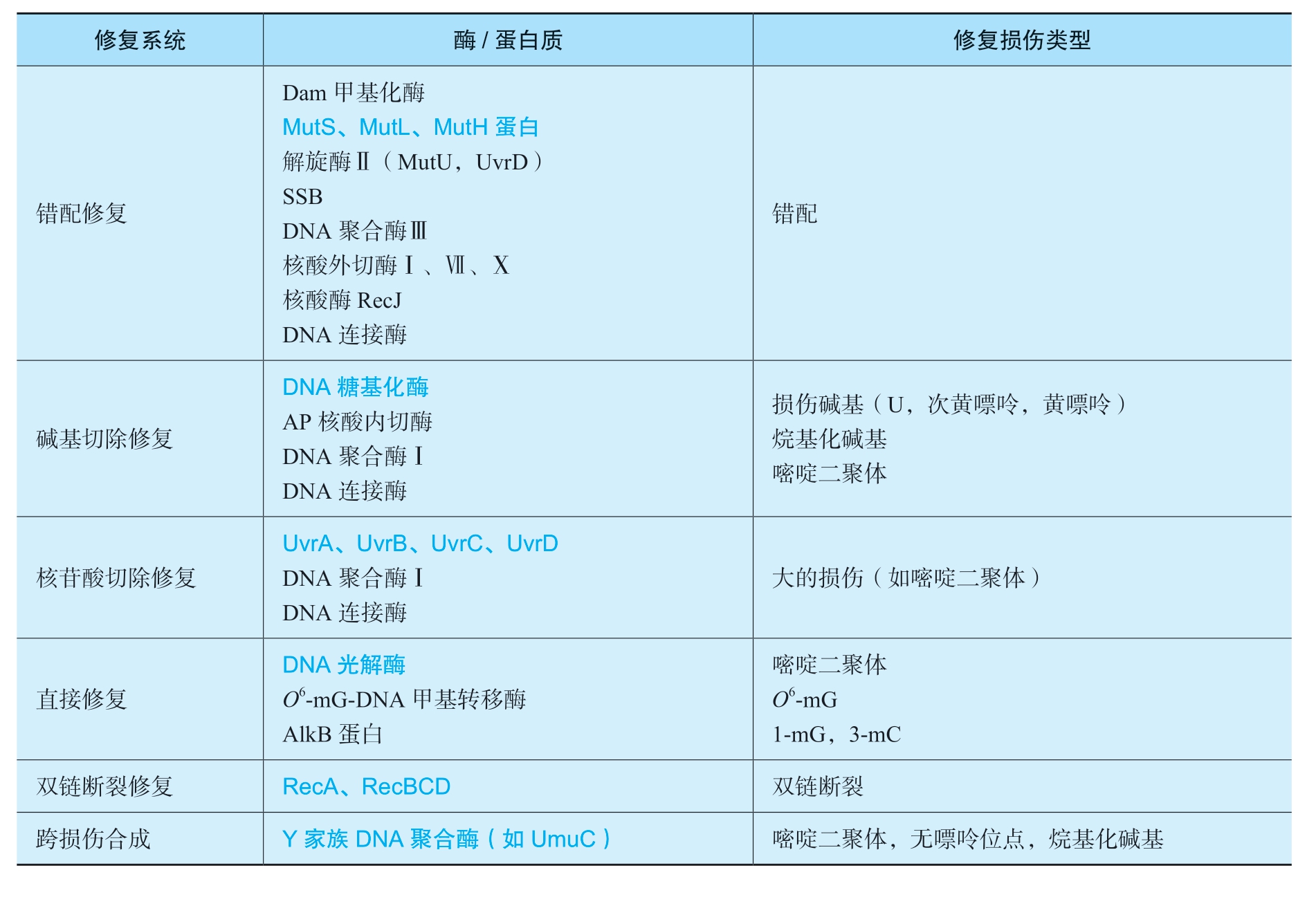
虽然DNA损伤导致的基因突变是生物进化的分子基础,但对个体而言绝大多数突变都是有害的。一个细胞只有两套甚至一套基因组DNA,并且DNA分子本身是不可替换的,所以一旦受到损伤必须及时修复,以维持遗传信息的稳定性和完整性。目前研究得比较清楚的DNA修复机制有错配修复、直接修复、切除修复、重组修复和SOS修复等(表2-6)。
表2-6 大肠杆菌DNA修复系统
● 在阐明DNA修复机制方面做出贡献的T. Lindahl(碱基切除修复)、P. Modrich(错配修复)和A. Sancar(核苷酸切除修复)获得2015年诺贝尔化学奖。
考虑到DNA复制错配率(10 -6 )及我们一生中细胞分裂次数(10 16 ),则一生中我们基因组(3×10 9 bp)的每个碱基对平均至少会发生2次自发突变。如果再考虑到其他因素诱发突变,在离世之际我们的基因组当面目全非了。当然事实并非如此,这要感谢DNA修复系统。典型哺乳动物的DNA损伤有99.9%都可及时修复(不能修复或未及时修复而成为突变的不到1/1000),最终将错配率降至10 -11 ~10 -10 。
DNA修复系统的多样性反映出DNA损伤的多样性和DNA修复的重要性。某些常见损伤类型如嘧啶二聚体可以通过几种修复系统修复。人类基因组有近200种基因编码产物组成修复系统。在很多情况下,其中任何一种编码产物功能缺失都会导致基因组不稳定性增加,肿瘤发生的可能性增加。
错配修复 (mismatch repair,MMR)是指在DNA复制完成后,在模板序列的指导下对新生链上的错配、单股插入缺失进行修复。大肠杆菌错配修复系统可修复GATC序列两翼1kb以内的错配,将复制精确度提高10 2 ~10 3 倍。大肠杆菌参与错配修复的蛋白质至少有12种(表2-6),其功能是识别模板或修复错配。
1.模板识别 错配修复的关键是识别子代DNA的模板链和新生链,然后才可根据模板序列修复新生链错配。真核生物和大多数细菌错配修复系统模板识别机制尚未阐明,仅大肠杆菌等个别细菌已经阐明。大肠杆菌亲代DNA两股链中GATC序列的A均甲基化为 N 6 -甲基腺嘌呤(m 6 A),称为 全甲基化GATC 。复制合成的子代DNA中,仅模板链GATC序列中的A甲基化,新生链未甲基化,称为 半甲基化GATC (图2-31)。
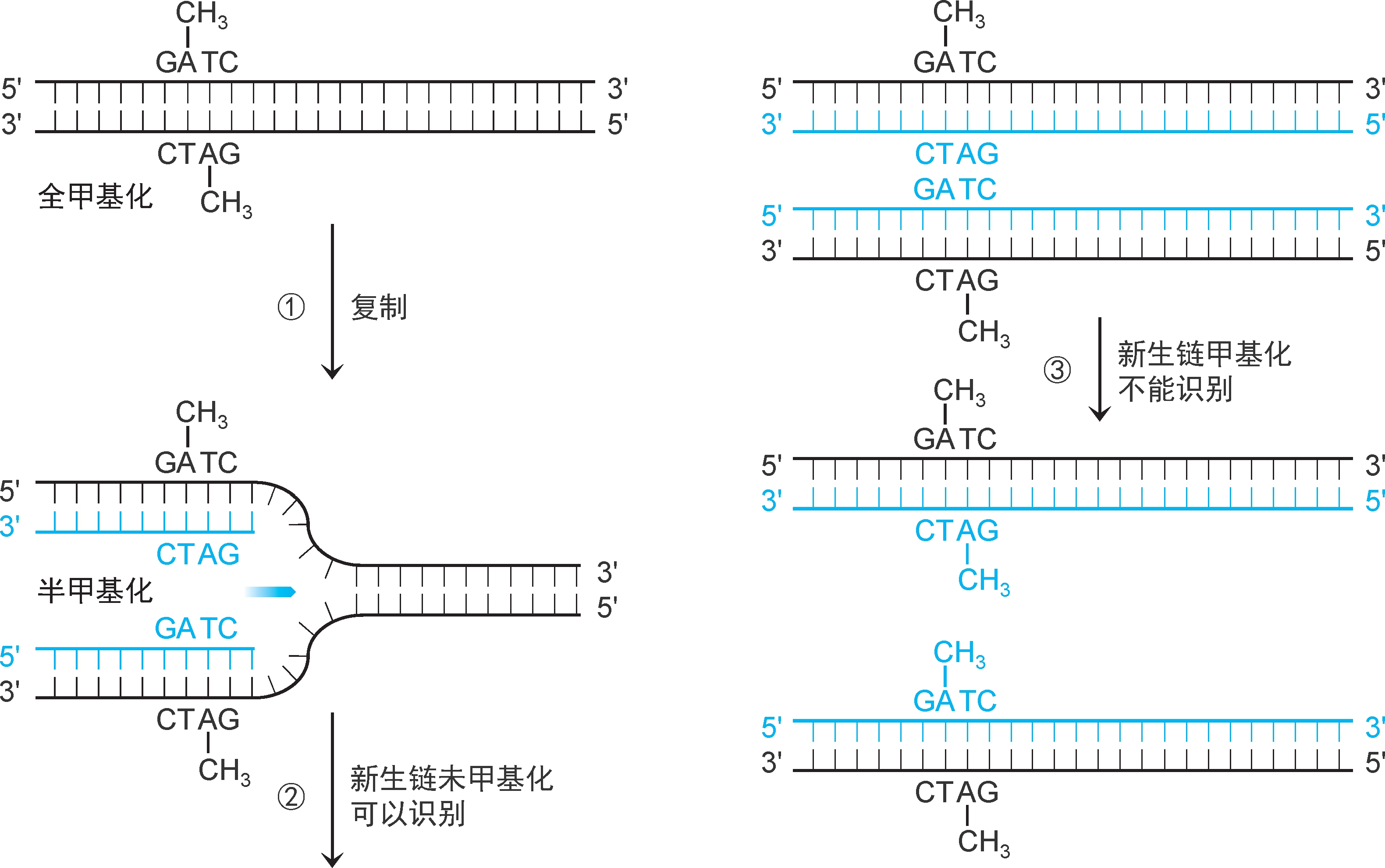
图2-31 GATC半甲基化和全甲基化
大肠杆菌错配修复蛋白MutS通过寻找半甲基化GATC识别模板链和新生链。①错配修复蛋白MutS二聚体扫描DNA(消耗ATP),结合于错配碱基对,募集错配修复蛋白MutL二聚体,形成 MutS- MutL复合物 。该复合物可以与除C-C之外的任何错配碱基对结合。②MutS-MutL复合物在错配碱基对两翼滑动扫描,寻找较近的一个半甲基化GATC(MutL消耗ATP),形成DNA环。③MutL募集错配修复蛋白MutH形成MutSLH复合物,激活MutH。MutH蛋白是一种 位点特异性核酸内切酶 ,催化半甲基化GATC新生链G的5'端磷酸二酯键水解,形成切口(pN-pGpApTpC,图2-32)。
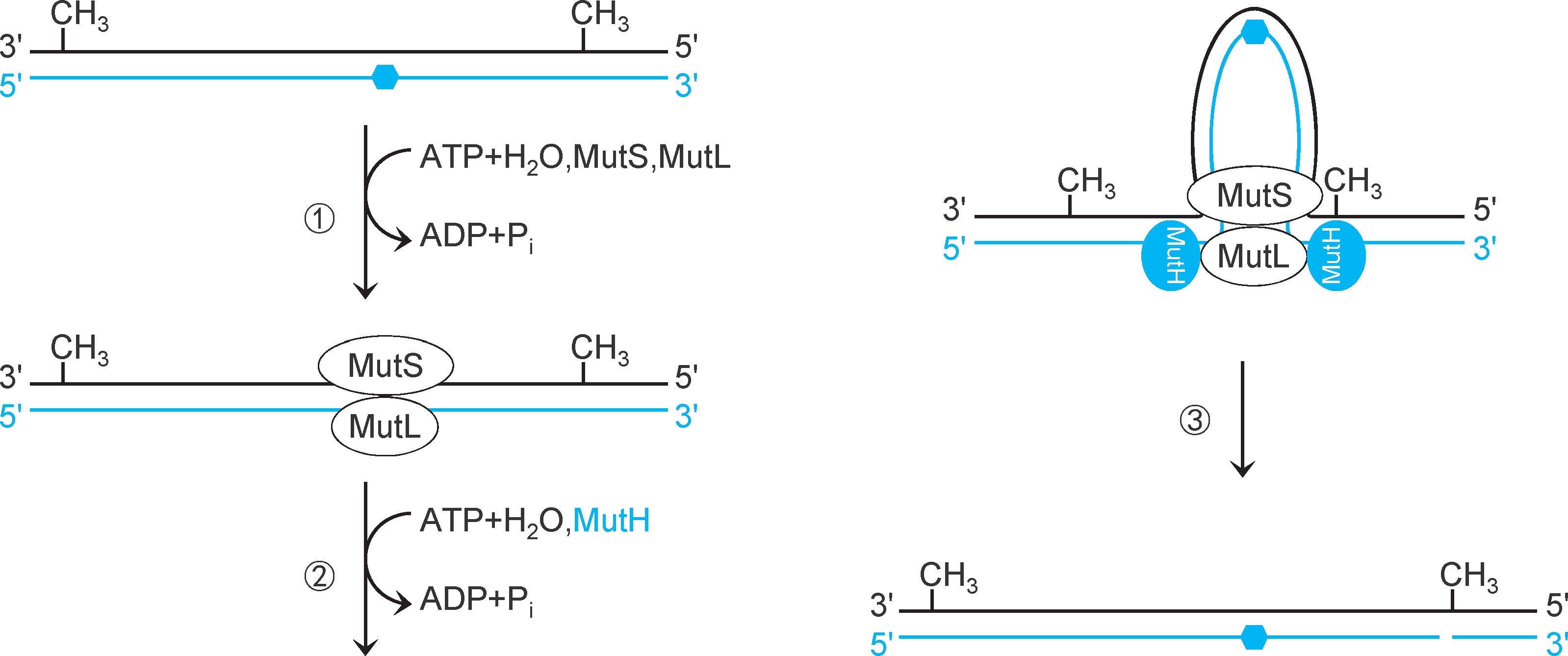
图2-32 大肠杆菌错配扫描
MutS-MutL复合物扫描范围覆盖错配碱基对两翼各1000bp。考虑到DNA中相邻GATC序列的平均距离为256bp(4 4 ),因此MutS-MutL复合物在错配碱基对侧翼会扫描到半甲基化GATC序列。
2.错配修复机制 ①解旋酶Ⅱ(又称UvrD解旋酶、解旋酶MutU,有ATPase活性和解旋酶活性,参与错配修复和核苷酸切除修复)从切口处向错配方向解旋DNA,SSB稳定,相应的核酸外切酶RecJ、ExoⅦ(5'→3'方向)或ExoⅠ、Ⅶ、Ⅹ(3'→5'方向)降解错配新生链,形成缺口。②DNA聚合酶Ⅲ催化合成DNA填补缺口,DNA连接酶催化封闭切口(图2-33)。
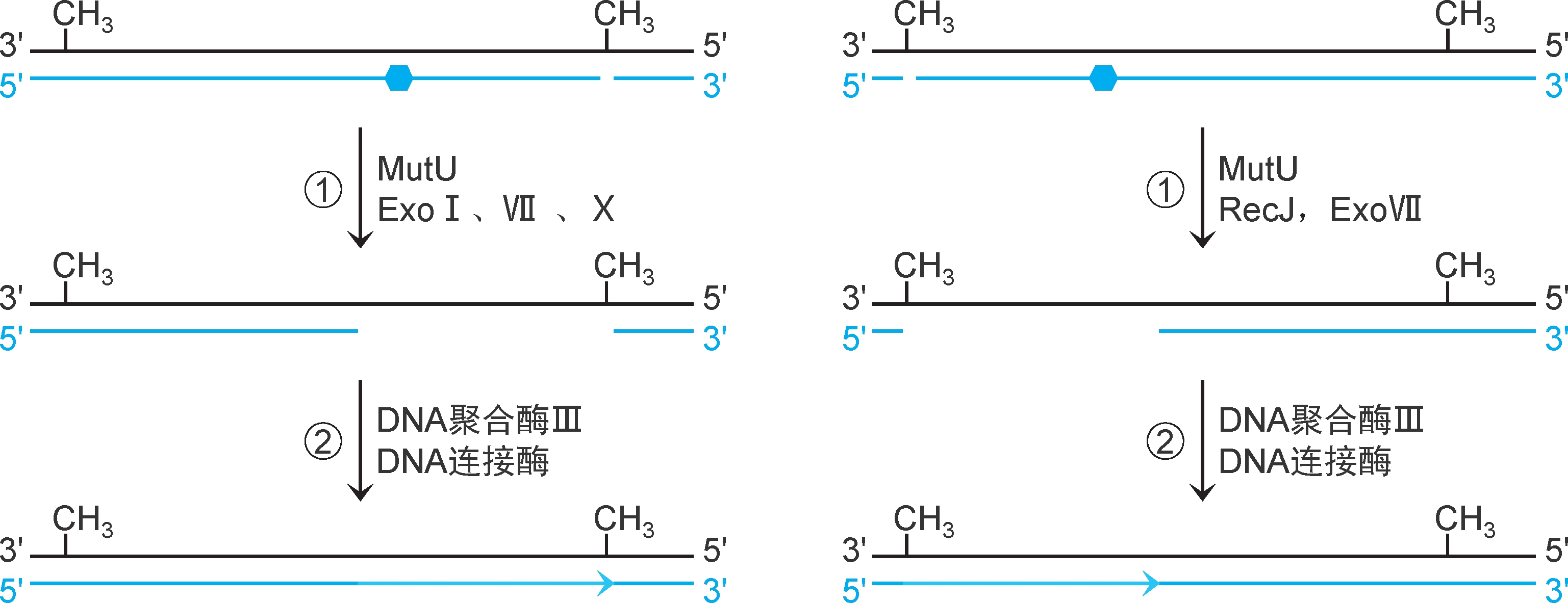
图2-33 大肠杆菌错配修复
错配修复要求子代DNA的GATC处于半甲基化状态,这一状态只能维持几秒至几分钟,之后将被甲基化为全甲基化状态。甲基化由大肠杆菌 Dam甲基化酶 催化(图2-31)。
注意:①错配修复极度耗能。错配碱基对与GATC序列可相隔1000bp,降解后至少需要消耗同样多dNTP中2倍量的高能磷酸基团,仅仅为了修复一个错配碱基。这从一个侧面提示维护基因组DNA完整性至关重要。②一旦半甲基化GATC序列被甲基化为全甲基化序列,将无法识别模板链和新生链,即使可以修复错配,修复率也只有50%。
真核生物错配修复系统有几种错配修复蛋白的结构和功能与细菌MutS、MutL类似,从酵母到人,真核生物最重要的MutS同源蛋白是MSH2、MSH3、MSH6。MSH2-MSH6异二聚体主要识别并结合单一错配碱基对,对较长错配结合力较弱。较长错配(2~6bp)由MSH2-MSH3识别并结合,或由两种二聚体串联识别并结合。MutL同源蛋白主要是MLH1-PMS1异二聚体,与MSH复合物结合并稳定之。真核生物新生链识别机制尚未阐明,但已明确与GATC序列无关。修复机制则与大肠杆菌类似。真核生物错配修复系统还可修复复制滑动导致的插入缺失,需要DNA聚合酶δ参与。人体这几种错配修复蛋白基因突变与某些常见的遗传性肿瘤易感综合征相关,进一步佐证了DNA修复系统的重要性。
许多真核生物有一种特别的修复系统,可把G-T配对修复为G-C碱基对而不是A-T碱基对,考虑到G-T中的T主要来自5mC脱氨基,这一修复系统有特殊意义。真核生物有5%C甲基化,5mC可自发脱氨基,故G-T是常见错配,且T为错配碱基。
切除修复 (excision repair)是指切除双链DNA的单链损伤,然后以正常互补链为模板,合成DNA填补缺口,将其修复。切除修复是细胞内最普遍的修复机制。原核生物和真核生物都有核苷酸切除修复系统和碱基切除修复系统,且均以核苷酸切除修复系统为主。两套系统都包括两个步骤:①由特异性核酸酶寻找损伤部位,切除损伤片段。②合成DNA填补缺口。
1.核苷酸切除修复(nucleotide excision repair,NER) 当嘧啶二聚体、烷基化碱基、碱基加成物(如苯并芘-鸟嘌呤)等DNA损伤导致双螺旋结构异常扭曲时,通常由核苷酸切除修复系统修复。核苷酸切除修复系统的关键酶是一种称为 切除核酸酶 (excinuclease)的多酶复合体。大肠杆菌的切除核酸酶为UvrABC,又称UvrABC修复系统,由UvrA、UvrB、UvrC构成(表2-7)。它不同于一般的核酸内切酶,可以同时水解损伤位点5'侧翼(上游)的第8个磷酸二酯键和3'侧翼(下游)的第5个磷酸二酯键(消耗ATP),释放损伤DNA片段,从而形成一个12~13nt(含1~2个损伤碱基)的缺口。真核生物切除核酸酶与大肠杆菌的基本功能一样,但特异性不同,是同时水解损伤位点5'侧翼的第22个磷酸二酯键和3'侧翼的第6个磷酸二酯键,从而形成一个27~29nt的缺口。
表2-7 大肠杆菌K-12株切除核酸酶UvrABC

(1)大肠杆菌核苷酸切除修复机制:①UvrA 2 B 2 扫描至损伤位点(消耗ATP),UvrA 2 脱离,UvrB 2 催化损伤位点解链,并募集UvrC,水解损伤位点侧翼特定磷酸二酯键,形成两个切口。②UvrD(即解旋酶Ⅱ)结合,沿3'→5'方向解链,释放损伤片段,形成12~13nt的缺口。③DNA聚合酶Ⅰ以互补链为模板,催化合成DNA填补缺口,DNA连接酶催化封闭切口(图2-34)。
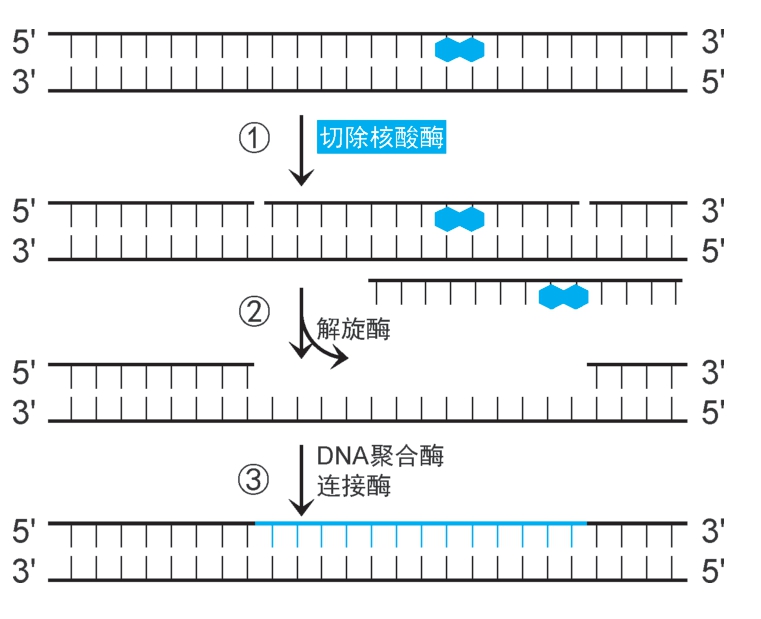
图2-34 核苷酸切除修复
(2)真核生物核苷酸切除修复机制:与大肠杆菌类似,只是参与修复的酶及其他蛋白质更多(至少25种),修复机制更复杂,有基因组修复和转录偶联修复两种机制。① 基因组修复 (global genome repair):修复系统包括8种基因编码的XPA、XPC、XPD、XPF、XPG、ERCC1等,其中XPC功能相当于UvrA,可以识别损伤并启动修复。此外,识别嘧啶二聚体等损伤还需要DNA损伤结合复合体(DDB)协助。② 转录偶联修复 (transcription-coupled repair,TCR):修复系统包括XPB、XPD、XPG、转录因子TFⅡH(第三章,85页)等,可修复转录过程中遇到的损伤,而且优先修复模板链。损伤导致RNA聚合酶Ⅱ变构,进而导致转录中止,而RNA聚合酶Ⅱ大亚基被降解。原核生物也有转录偶联修复系统。
真核生物两种修复机制都需要由通用转录因子TFⅡH在损伤位点解链约20bp。解旋酶XPB是TFⅡH的一个亚基(TFⅡH p89),其功能是参与转录启动子解链及损伤位点3'→5'解链。解旋酶XPD也是TFⅡH的一个亚基(TFⅡH p80),其功能是稳定转录起始复合物及参与损伤位点5'→3'解链(相当于UvrB),之后由ERCC1-XPF、XPG分别从损伤位点5'侧翼、3'侧翼切开,形成27~29nt的缺口,由DNA聚合酶δ或ε催化合成DNA片段填补。
2.碱基切除修复(base excision repair,BER) 可以修复DNA的单碱基损伤,即依次由DNA糖基化酶催化切除损伤碱基,形成AP位点,AP核酸内切酶催化切断AP位点磷酸酯键,外切酶切除脱氧核糖,DNA聚合酶催化加接核苷酸,DNA连接酶催化封闭切口。
(1)大肠杆菌碱基切除修复机制:大肠杆菌碱基切除修复酶系包括五类酶。① DNA糖基化酶 (DNA glycosylase,DNA糖苷酶,DNA glycosidase):有几十种,每一种都特异识别并水解一种损伤碱基形成的糖苷键,释放损伤碱基,形成AP位点。可分为单功能糖基化酶(催化水解释放损伤碱基,形成AP位点)和双功能糖基化酶(除催化释放损伤碱基形成AP位点外,还有AP裂解酶活性)。② AP核酸内切酶 (AP endonuclease,AP裂合酶):有十几种,不同AP核酸内切酶催化机制及专一性不同。可分为水解酶(催化AP位点上游磷酸酯键水解,形成3'端为羟基、5'端为5'-磷酸脱氧核糖的切口)和裂解酶(催化AP位点下游磷酸酯键裂解——β消除,形成3'端为去饱和醛、5'端为5'-磷酸脱氧核苷酸的切口)。③ 核酸外切酶 :催化脱去5'-磷酸脱氧核苷酸,如RecJ。④DNA聚合酶Ⅰ:催化加接一个脱氧核苷酸(图2-35左)。在无核酸外切酶时,可通过切口平移合成一段寡脱氧核苷酸(有时简称寡核苷酸)(图2-35右)。⑤DNA连接酶。
(2)真核生物碱基切除修复机制:真核生物有两条碱基切除修复途径,均在DNA糖基化酶(人类基因组编码的11种DNA糖基化酶已被鉴定)切除损伤碱基后由AP核酸内切酶APE1(还有3'→5'外切酶活性)催化AP位点上游的磷酸酯键水解,形成3'端为羟基、5'端为5'-磷酸脱氧核糖的切口,再由FEN1切除AP位点5'-磷酸脱氧核糖甚至1~9nt脱氧核苷酸:① 短修补途径 (SP-BER,单核苷酸碱基切除修复,SN-BER):APE1募集DNA聚合酶β,催化切除AP位点5'-磷酸脱氧核糖,加接一个脱氧核苷酸,DNA连接酶1或3催化封闭切口(图2-35左)。② 长修补途径 (LP-BER):APE1募集PCNA。PCNA募集FEN1,催化切除AP位点5'-磷酸脱氧核糖及1~9nt脱氧核苷酸,PCNA募集DNA聚合酶δ或ε等,催化加接2~10nt脱氧核苷酸。DNA连接酶1催化封闭切口(图2-35右)。

图2-35 碱基切除修复
● 鸟嘌呤会被氧化成8-氧鸟嘌呤(8-oxoG)。8-oxo-dGTP在DNA复制时会以同样几率与A或C配对。大肠杆菌在三个环节修复8-oxoG:①底物环节:dGTP被氧化成8-oxo-dGTP时,8-oxodGTP酶MutT催化8-oxo-dGTP水解,防止其掺入DNA:8-oxo-dGTP+H 2 O→8-oxo-dGMP+PP i ;②产物环节:DNA中的dGMP被氧化成8-oxo-dGMP时,DNA糖基化酶MutM催化8-oxo-dGMP水解,启动碱基切除修复;③复制环节:DNA复制过程中有dAMP掺入与8-oxo-dGMP互补配对时,DNA糖基化酶MutY催化dAMP水解,启动碱基切除修复。
直接修复 (direct repair)是指不切除损伤碱基或核苷酸,直接将其修复,例如嘧啶二聚体的光修复和烷基化碱基的去烷基化修复。
1.光修复 嘧啶二聚体有多种修复机制,其中由A. Sancar团队阐明、由DNA光解酶催化进行的光修复是高度特异的直接修复方式。大肠杆菌 DNA光解酶 (DNA photolyase,又称DNA光裂合酶,每个细胞有10~20分子)以FADH - 、 N 5 , N 10 -次甲基四氢叶酰多谷氨酸为辅助因子(发色团),被300~600nm光激活后可催化环丁烷嘧啶二聚体解聚。DNA光解酶广泛存在于低等单细胞生物到鸟类及各种植物,但除有袋动物之外的有胎盘哺乳动物没有此酶。人类基因组编码两种DNA光解酶同源蛋白,被称为隐花色素(cryptochrome),其中隐花色素1是生物钟(circadian clock)的核心成分。
2.去烷基化修复 有些酶可以识别DNA中的烷基化碱基。例如,①大肠杆菌 O 6 -甲基鸟嘌呤( O 6 MeG,m 6 G,与胸腺嘧啶配对,导致G→A转换)可被 O 6 -甲基鸟嘌呤-DNA甲基转移酶(MGMT,单体酶)识别,并且直接将其 O 6 -甲基转至MGMT的Cys139的巯基上(不可逆)。此外,该酶还可以同样机制转 O 4 -甲基胸腺嘧啶(m 4 T)的 O 4 -甲基。②3-甲基胞嘧啶(m 3 C)和1-甲基腺嘌呤(m 1 A)的甲基(甲基化发生在单链状态,影响配对)可以被氧化脱去,生成甲醛(以Fe 2+ 为辅助因子):α-酮戊二酸+ m 1 A-DNA+O 2 =A-DNA+CO 2 +甲醛+琥珀酸。原核生物(大肠杆菌AlkB)和真核生物均存在该修复酶类。RNA也有该修复机制。
重组修复 (recombinational repair)系统可修复以下损伤:①复制进行至模板损伤(如嘧啶二聚体)部位时,复制体不能根据碱基配对原则合成新生链,会越过损伤继续复制,新生链对应模板损伤部位留下缺口(图2-36左)。②复制进行至模板切口或断裂位点时,复制体解体(collapse),复制叉一臂成为双链断裂末端(图2-37右)。两种损伤各由特定修复系统修复。
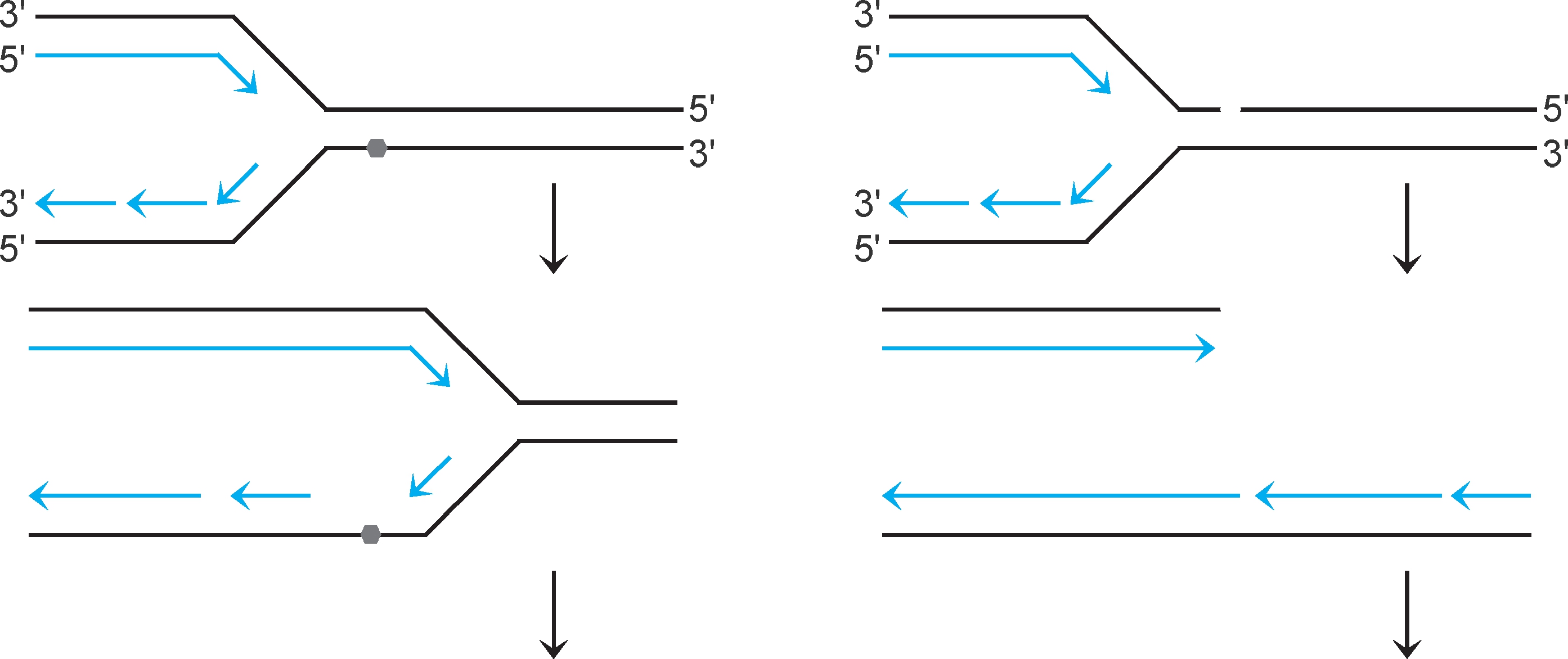
图2-36 影响复制的损伤
1.缺口修复 由 重组酶RecA 和核酸外切酶Ⅴ(又称 RecBCD复合体 ,为RecB、RecC、RecD三聚体结构,都有核酸酶、解旋酶、ATP酶活性,作用是提供3'黏性末端)等催化,从姐妹染色单体移植同源序列修补缺口(图2-37)。复制完成时,损伤并未修复,可以通过切除修复机制进行修复。
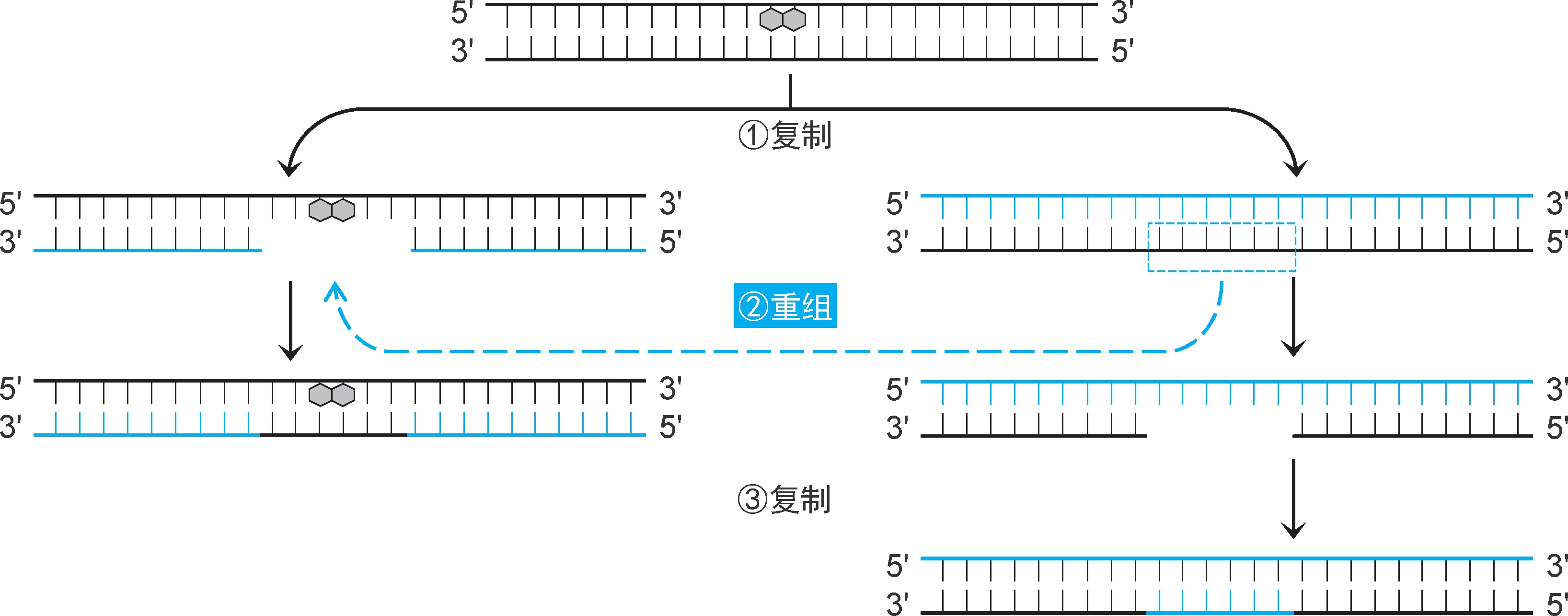
图2-37 重组修复
重组酶RecA和核酸外切酶Ⅴ还可在缺口形成前催化模板修复(图2-38):复制在损伤位点中止,复制体解体。①复制叉后退,前导链和后随链形成双链。②模板损伤进行切除修复。③复制叉恢复推进,复制体再形成,复制重启。此修复过程需要RecA稳定DNA单链,RecB、RecC参与切除修复,有时会有重组发生(图中未示)。
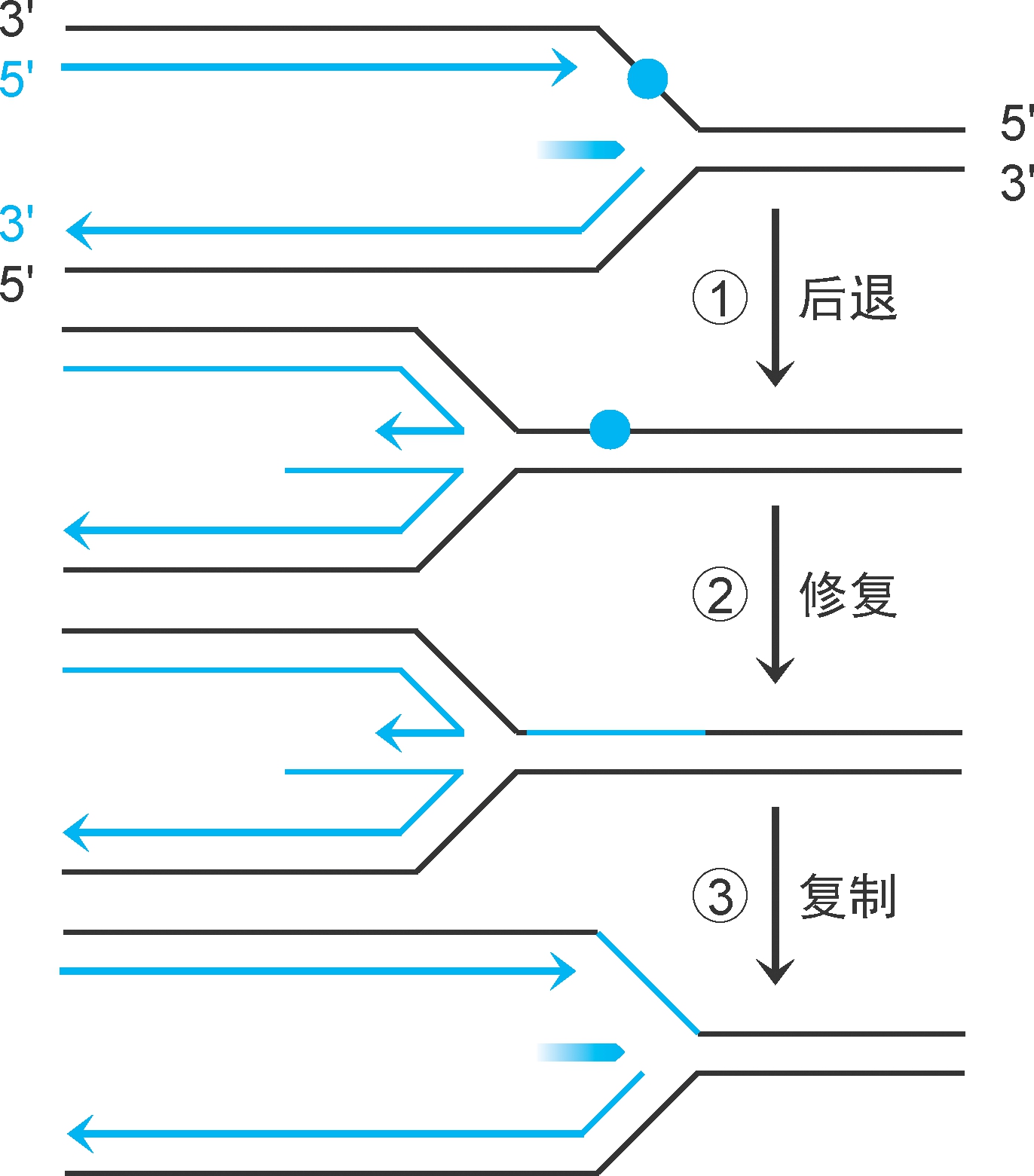
图2-38 复制中修复
2.双链断裂修复 包括同源重组修复和非同源末端连接。同源重组修复是酵母双链断裂的主要修复机制(图2-41,66页)。 非同源末端连接 (nonhomologous end joining,NHEJ)是将断裂末端经过适当加工重新连接,由J. Moore和J. Haber于1996年报道,是高等生物双链断裂的主要修复途径,机制如下:①断裂末端募集DNA修复蛋白XRCC6-XRCC5(Ku70-Ku80)二聚体(解旋酶Ⅱ),进而募集DNA-PK催化亚基形成DNA-PK复合体,募集5'→3'外切酶Artemis,形成断裂末端复合体,并二聚化。② 断裂末端复合体中DNA-PK催化Artemis磷酸化激活,呈现5'→3'内切酶活性。末端部分水解。③XRCC6-XRCC5催化断裂末端解链。④末端间单链局部退火。⑤Artemis切去游离末端,缺口由Pol μ或Pol λ催化合成填补,切口由DNA连接酶Ⅳ催化封闭(图2-39)。
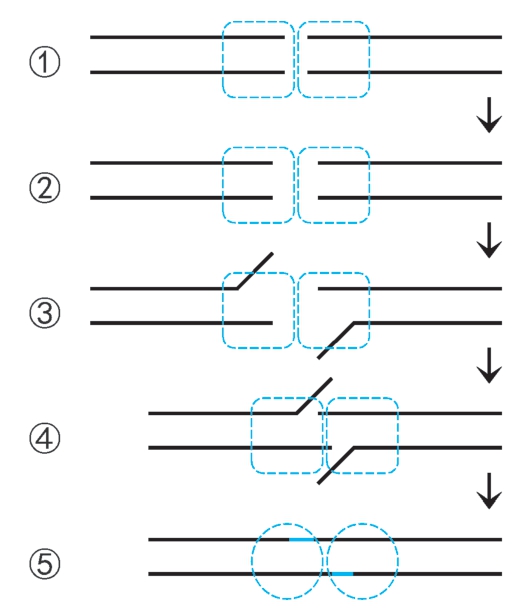
图2-39 非同源末端连接
NHEJ未保留原始序列,会在连接点引入突变,NHEJ体系缺陷个体肿瘤发生风险增加。
DNA链断裂监控与肿瘤:人体DNA断裂由聚ADP核糖聚合酶1(PARP-1,聚ADP核糖转移酶)监控。PARP-1缺失个体DNA修复能力低下,单链断裂积累,复制时形成双链断裂。某些类型的乳腺癌、卵巢癌存在双链断裂修复缺陷,即 BRCA1 、 BRCA2 或相关基因缺失。这些细胞一旦缺失PARP-1后果极其严重,因为单链断裂积累导致复制时染色体断裂。基于此开发的PARP-1抑制剂类抗肿瘤药物奥拉帕利(XL01XX)已经上市,可用于治疗存在双链断裂修复缺陷的肿瘤患者,例如用于某些 BRCA1 或 BRCA2 缺失型乳腺癌或卵巢癌患者的维持治疗。
DNA修复系统的修复能力与DNA的损伤程度相关。DNA损伤严重时会激活与DNA修复有关的一组基因,其中有许多参与DNA修复,这一现象称为 SOS反应 (SOS应答,SOS response),这组基因称为 SOS基因 (SOS gene)。SOS反应产生两类效应:①诱导切除修复和重组修复等修复系统基因的表达,从而提高修复能力。②启动SOS修复系统。SOS修复系统的基因通常处于沉默状态,紧急情况下才被整体激活,指导合成40多种 SOS蛋白 (SOS protein),激活机制见第五章(145页)。
大肠杆菌SOS修复系统的核心是DNA聚合酶Ⅳ(DinB)和DNA聚合酶Ⅴ(UmuCUmuD' 2 )。它们对碱基的识别能力差,能催化有损伤DNA模板的复制,称为 跨损伤合成 (translesion synthesis,TLS,跨损伤复制,translesion replication)。跨损伤合成不能校对,错配率高达10 -3 。
跨损伤合成是DNA损伤严重引起的一种应急反应,特点是保真性降低、突变率大增。跨损伤合成仅在复制叉推进过程中遇到DNA损伤而无法正常完成复制时才会启动(图2-36左)。其本质是DNA聚合酶Ⅳ和Ⅴ不严格执行碱基配对原则,而是近乎随机连接核苷酸。这种机制虽然使复制得以进行下去,但基本未修复损伤,最终发展为突变,造成突变积累,所以称为 SOS修复 (SOS repair,易错修复,error-prone repair)。
SOS修复虽然最终会导致一些细胞死亡,但毕竟使另一些细胞得以存活。这种以发生突变为代价的修复似为无奈之举,但对存活突变体而言是值得的。
人体有5种DNA聚合酶催化跨损伤合成(其中4种属于DNA聚合酶Y家族),并且它们有一定的校对功能。例如,真核生物都有DNA聚合酶η,它催化胸腺嘧啶二聚体的跨损伤合成时极少发生错配,因为它恰好优先选择连接腺苷酸。
DNA损伤后果取决于DNA的损伤程度和细胞的DNA修复能力。如果细胞不能修复DNA,就会因基因功能异常而导致疾病。一些遗传病和肿瘤等就与DNA修复缺陷有关。
1.着色性干皮病 是一种常染色体隐性遗传病,患者存在DNA修复缺陷,特别是核苷酸切除修复缺陷,编码核苷酸切除修复系统的8个基因中(特别是 XPA 、 XPB 、 XPC )有突变发生,不能修复由紫外线照射等引起的表皮细胞DNA损伤,特别是嘧啶二聚体,导致高突变率,是正常人的1000多倍。着色性干皮病的特征是患者对日光尤其是紫外线特别敏感,易被晒伤,皮肤暴露部分形成大量黑斑甚至溃烂,常在婴儿期即显皮肤症状:皮肤干燥,真皮萎缩、角质化,眼睑瘢痕化,角膜溃烂,学龄前即发展为基底细胞上皮瘤及其他皮肤癌,许多患者因皮肤癌转移而在30岁前死亡。
2.遗传性非息肉病性结直肠癌 又称Lynch综合征,约占全部结肠癌的2%。患者存在错配修复缺陷(约20%肿瘤患者存在错配修复缺陷),不能修复复制滑动导致的插入缺失,因而其微卫星DNA长度异常。编码错配修复系统因子的五个基因即 MLH1 (与大肠杆菌 mutL 同源)、 MSH2 (与大肠杆菌 mutS 同源)、 MSH6 、 PMS1 、 PMS2 中只要有一个基因(多为 MLH1 和 MSH2 )发生突变,就可能导致细胞错配修复缺陷,基因组稳定性得不到有效维护,调节细胞生长的基因容易发生突变,细胞容易发生恶性转化。缺陷个体通常早发肿瘤,以结肠癌最多。
3. Cockayne综合征 是一种罕见的隐性遗传病,一种早衰症(progeroid syndrome),患者存在与转录偶联的核苷酸切除修复缺陷,特征是发育迟缓、神经退行性疾病、老年貌,多在12岁前死于早衰。
4.乳腺癌 大多数卵巢癌和女性乳腺癌患者并无已知患癌倾向,但约10%的患者存在 BRCA1 或 BRCA2 缺陷。有 BRCA1 或 BRCA2 缺陷女性乳腺癌发生风险比正常女性高70%。人BRCA1和BRCA2与一组蛋白质共同参与转录、染色体维持、DNA修复、细胞周期调控。作用机制为参与双链断裂重组修复。