
下载掌阅APP,畅读海量书库
立即打开



通过典型案例分析及广泛的药物合成实践经验,可将药物合成工艺路线选择与改进的原则归纳为以下几点:①化学合成途径简捷、路线短;优选“汇聚式”。②原辅料品种少、安全低毒、价廉易得、供应稳定。③反应温和、易于控制;优选“平顶型”。④副反应少,中间体和产物易分离、纯化;优选“一勺烩”。⑤“三废”少且易处理,“原子经济性”好。⑥设备条件要求不苛刻,推进现代化制药设备的使用。⑦针对手性药物,充分开发不对称催化合成及手性拆分工艺。⑧收率最佳、成本最低、经济效益好。这些基本原则是工艺路线科学抉择的具体体现,对工艺路线的评价、选择与改进有指导意义。在这里对这几方面问题依次分析,出发点仍是如何找到理想的药物合成工业生产路线。
化学合成途径简捷、路线短;优选“汇聚式”。
在技术路线理论可行的基础上,一条设计巧妙、反应步骤少的路线,往往具有总收率高、合成周期短、易于控制、易于处理、“三废”少、成本低等特点,为工业生产带来巨大的经济效益,实现了环境影响最小化。因此,合成路线短是评价一条路线最为简单和直观的标准。同时,缩短合成路线也是工艺路线改进的主要手段,是现代化制药、绿色制药的本质化、高效化的核心突破点。
托品酮(Tropinone,4-44)的合成就是很好的例子,如图4-16,托品酮最早是由德国化学家Willstätter(维尔施泰特)在1902年经过15步全合成完成的,但由于步骤较多,总产率只有0.75%。对这一成功,在当时是实验室全合成复杂天然产物的重要事件之一,标志着全合成方法的诞生。1917年,应该化学家罗伯特·鲁宾逊(Robinson)创造了简短的托品酮合成法,该法是有机合成中的经典路线之一,仅以结构简单的丁二醛、甲胺和3-氧代戊二酸为原料(4-43),在仿生条件下,利用Mannich(曼尼希)反应,仅通过三步反应(一锅反应)就合成了托品酮,而且产率达到17%,后经改进,收率可以提高至90%以上。
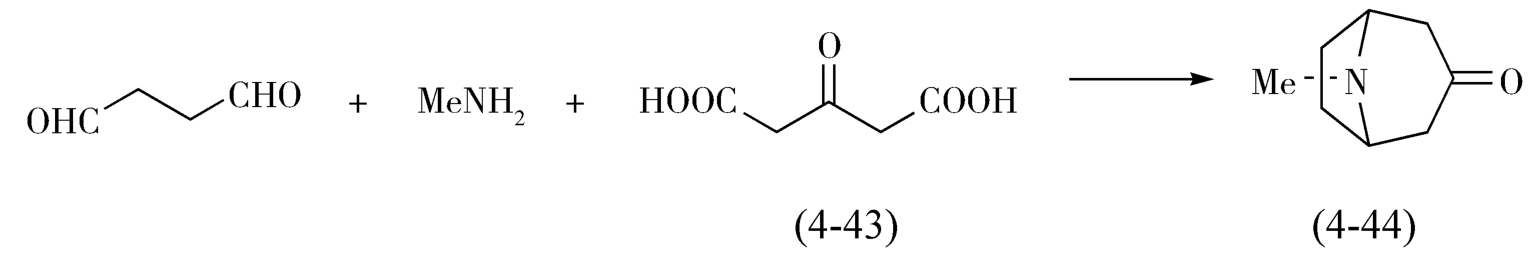
图4-16 三步法合成托品酮的路线图
艾瑞昔布(Imrecoxib,4-45)的合成也能充分体现缩短路线步骤带来的工艺优化。艾瑞昔布是环氧合酶抑制剂,具有优良的抗炎、镇痛作用,用于类风湿关节炎、骨关节炎等。早期报道的合成路线以对甲磺酰基溴代苯乙酮(4-46)为起始原料,依次经过还原、 N -烷基化、酰化、氧化和缩合共5步反应合成得到艾瑞昔布(4-45),如图4-17所示。随后,研究人员在上述路线的基础上又设计了“两步法”路线,如图4-18,仍以对甲磺酰基溴代苯乙酮(4-46)为起始原料,在三氯化铝的催化下先与对基苯乙酸发生 O -烷基化反应,随后直接进行分子内的缩合反应得到内酯中间体(4-47),内酯在醋酸的作用下,与丙胺发生氨解、取代反应,完成艾瑞昔布(4-45)的制备。与前述的五步法路线相比,两步反路线在技术层面的优势十分明显:①路线明显缩短。②两步法是更为典型的汇聚式合成策略。③两种主要原料并未改变,以对甲基苯乙酸替代相应酰氯,避免了硼氢化钠、Jones试剂的使用,原料、试剂种类更少、价格更廉。④避免了还原、氧化等过程,规避了强碱、强氧化和高度无水等反应条件,使反应过程更温和、更可靠、更安全。⑤后处理过程明显简化,产物纯度良好。
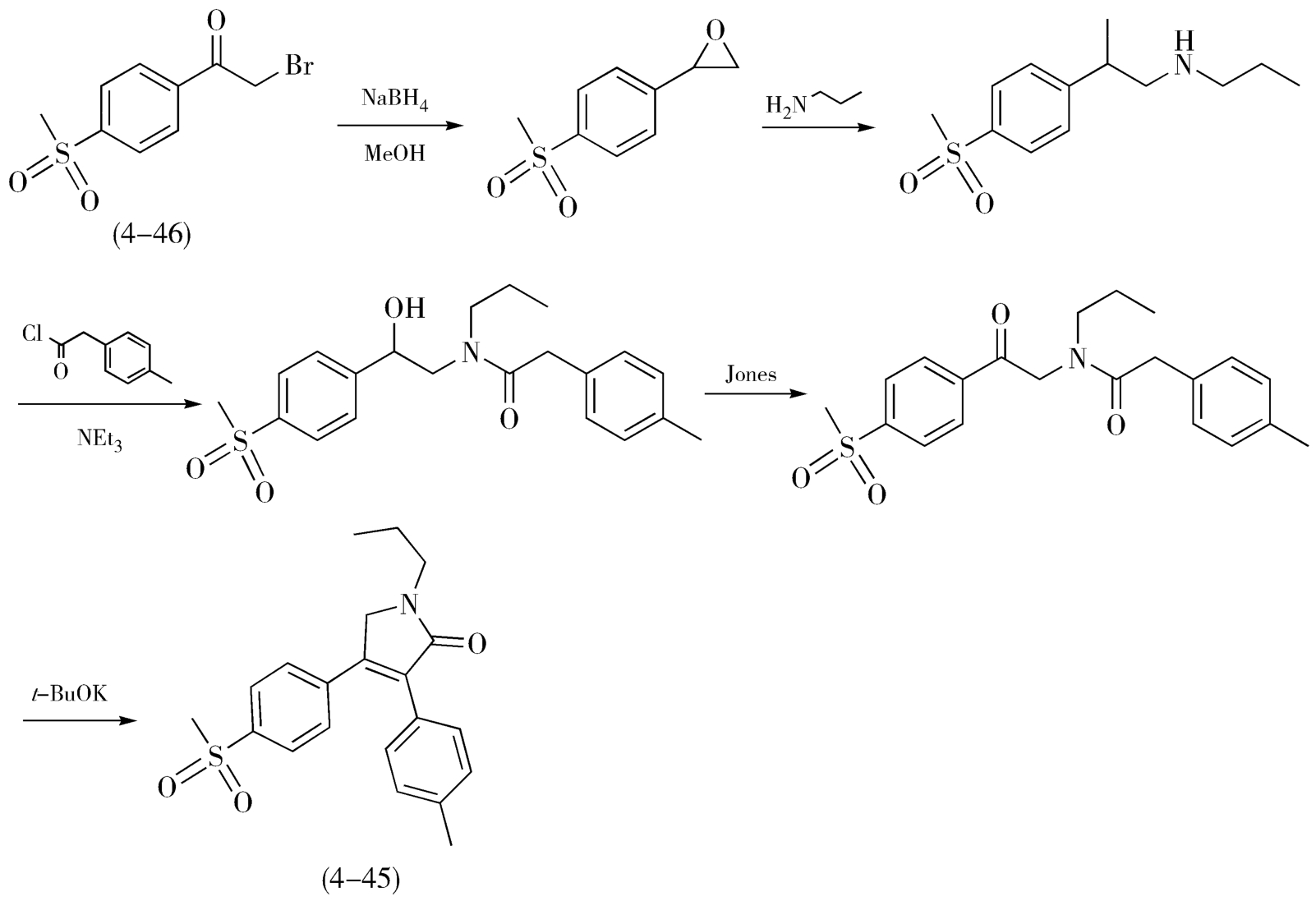
图4-17 艾瑞昔布的“五步法”合成路线图
布洛芬由广泛采用的6步Boots法改进为3步的BHC法,也充分体现了步骤少带来的显著优势;紫杉醇由二十多步的全合成到4步的半合成的路线改进,代表了对天然活性物质的全合成到半合成的整体途径的改进模式,也充分说明了合成路线短带来的进步和工业开发价值。
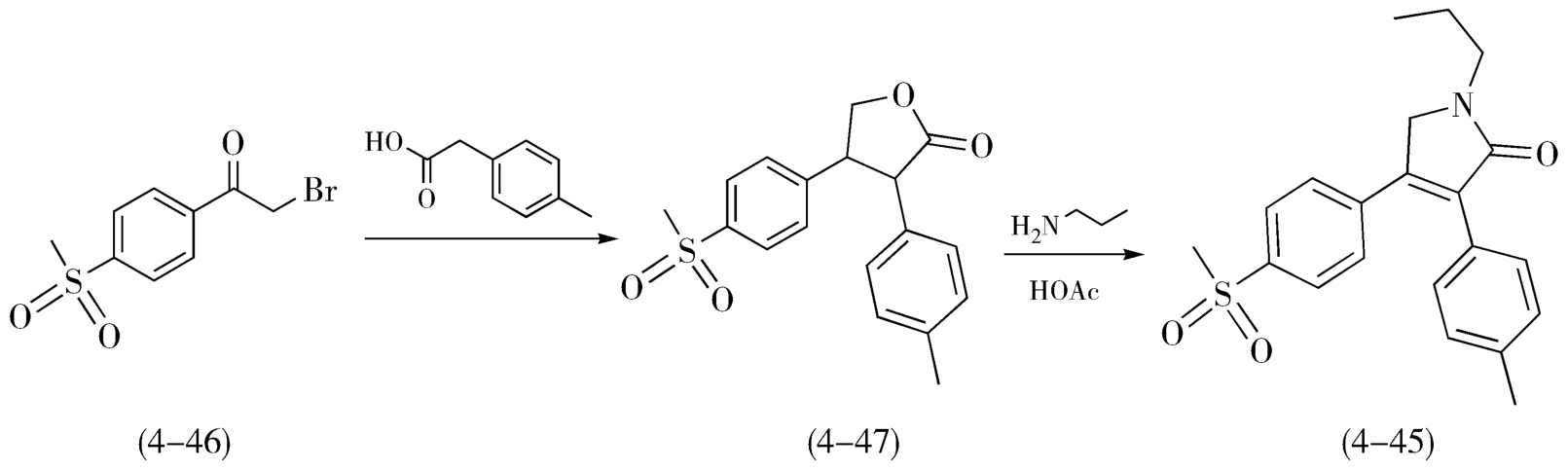
图4-18 艾瑞昔布的“两步法”合成路线图
合成路线尽可能短,是评价工艺路线最直观的原则。但是,不一定最短的路线就是最简捷的,还应考虑反应条件实施难度、后处理繁杂程度等其他方面的问题。比如,第二章阐述克霉唑的合成路线,路线一和路线二都是3步反应,可最后工业化广泛采用的是有5步反应的路线三,可以看出,路线的选择,除了步骤长短,还有考虑原辅材料、反应条件、后处理等等问题。若简短的路线存在后处理复杂、总收率低、安全性差等问题,也不防多做一两步,选择整体路径简单易行、途径简捷的稍长路线。
合成路线选择时,还要考虑合成路线构建方式问题,即合成装配方式。对于一个多步骤的合成路线而言,有两种典型的装配方式,一种是“直线式”,即一步一步的进行反应,每一步增加目标分子的一个新单元,最后构建整个分子,如图4-19所示;另一种是“汇聚式”,即分别构建目标分子的主要部分,几个中间体再连接到一起,最终得到新分子,如图4-20所示。汇聚式合成法具有一定的优势:①风险小:生产过程中一旦出现差错,损失某个中间体,也不会低整条路线,造成毁灭性影响。②总收率高:假定各步收率相同、步数相同的两条路线,“汇聚式”总收率明显要比“直线式”高。③规模小、成本小:汇聚式总收率更高,即所需要的起始原料和所有试剂、耗材的用量大大减小,节约了试剂成本。④规模小:反应容器和其他设备也相对较小,增加了设备使用的灵活性,降低了设备成本和操控成本。⑤合成周期也较低,时间成本低。
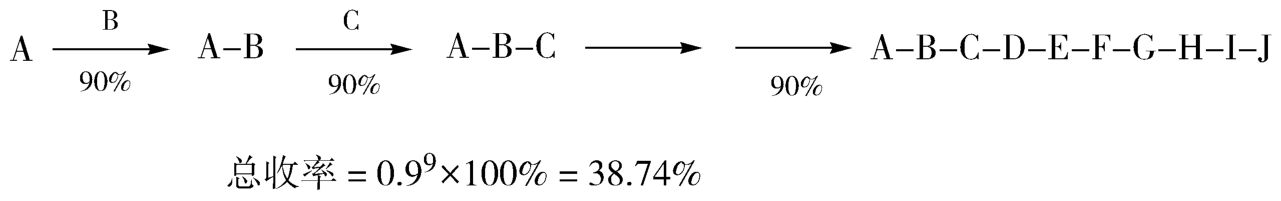
图4-19 “直线式”装配方式及收率示意图
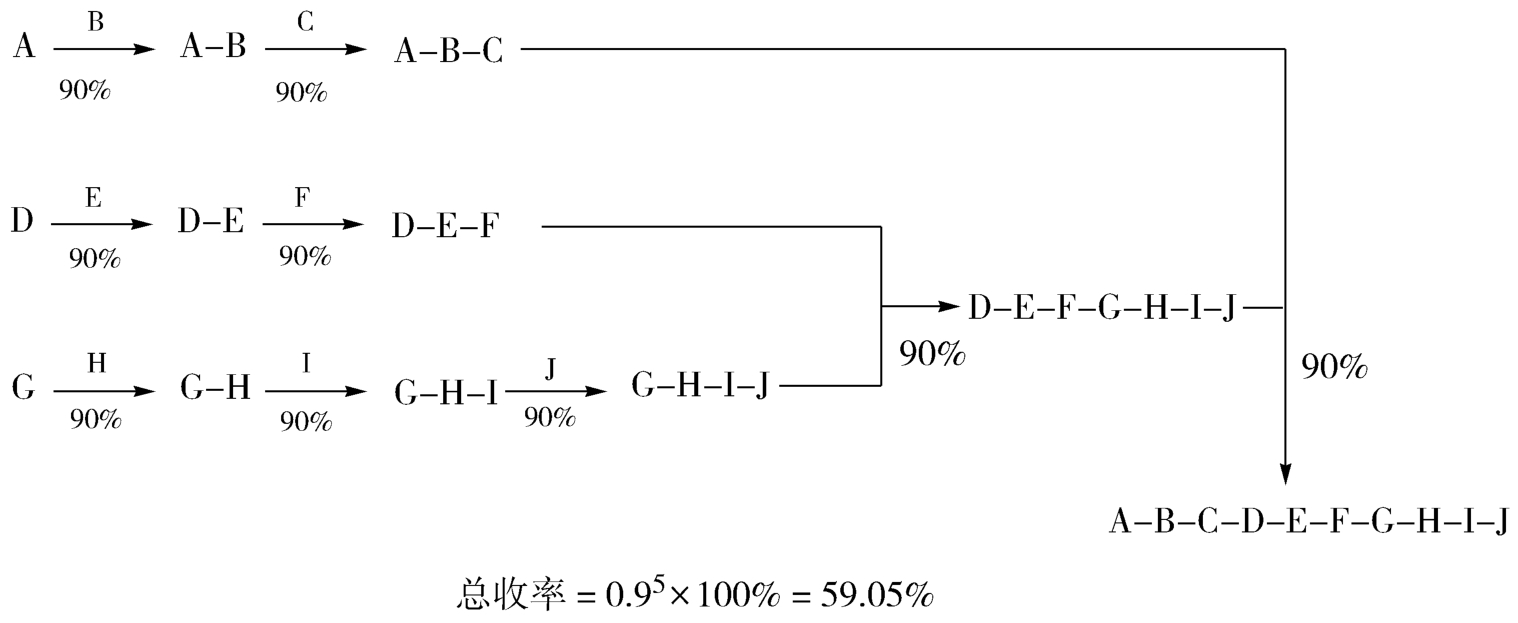
图4-20 “汇聚式”装配方式及收率示意图
利奈唑胺(Linezolid,4-48)是用于抗感染的χ唑类抗生素,由美国Pharmacia&Upjohn公司研制并于2000年上市。利奈唑胺的合成采用了“直线式”路线,如图4-21,以3,4-二硝基苯(4-49)为原料,与吗啉反应后再经氢化还原得到3-氟-4-吗啉基苯胺(4-50),所得苯胺进一步与氯甲酸苄酯反应得化合物(4-51),然后在丁基锂的作用下与化合物(4-52)反应制得含恶唑烷酮环中间体(4-53),恶唑烷酮环中间体(4-53)上的羟基经磺酰基保护、叠氮化取代、加氢还原后,转变为目标分子利奈唑胺(4-48)。随后,研究人员根据利奈唑胺的结构特点,开发出了多种采用“汇聚式”合成的工艺路线,如图4-22所示,在按照“直线式”路线中合成化合物(4-51)的同时,以右旋环氧氯丙烷(4-55)为原料,经成盐反应、乙酰化反应、制备中间体( S )- N -(3-氟-2-羟丙基)乙酰胺(4-56),然后将这两个片段在碱性条件下结合制得利奈唑胺(4-48)。对比上述两条合成路线,步骤总数相当,但采用汇聚式的合成路线更具优势,可同时进行两个中间片段的制备,在降低风险的同时,大大节约产物的合成周期、降低了规模和总成本。
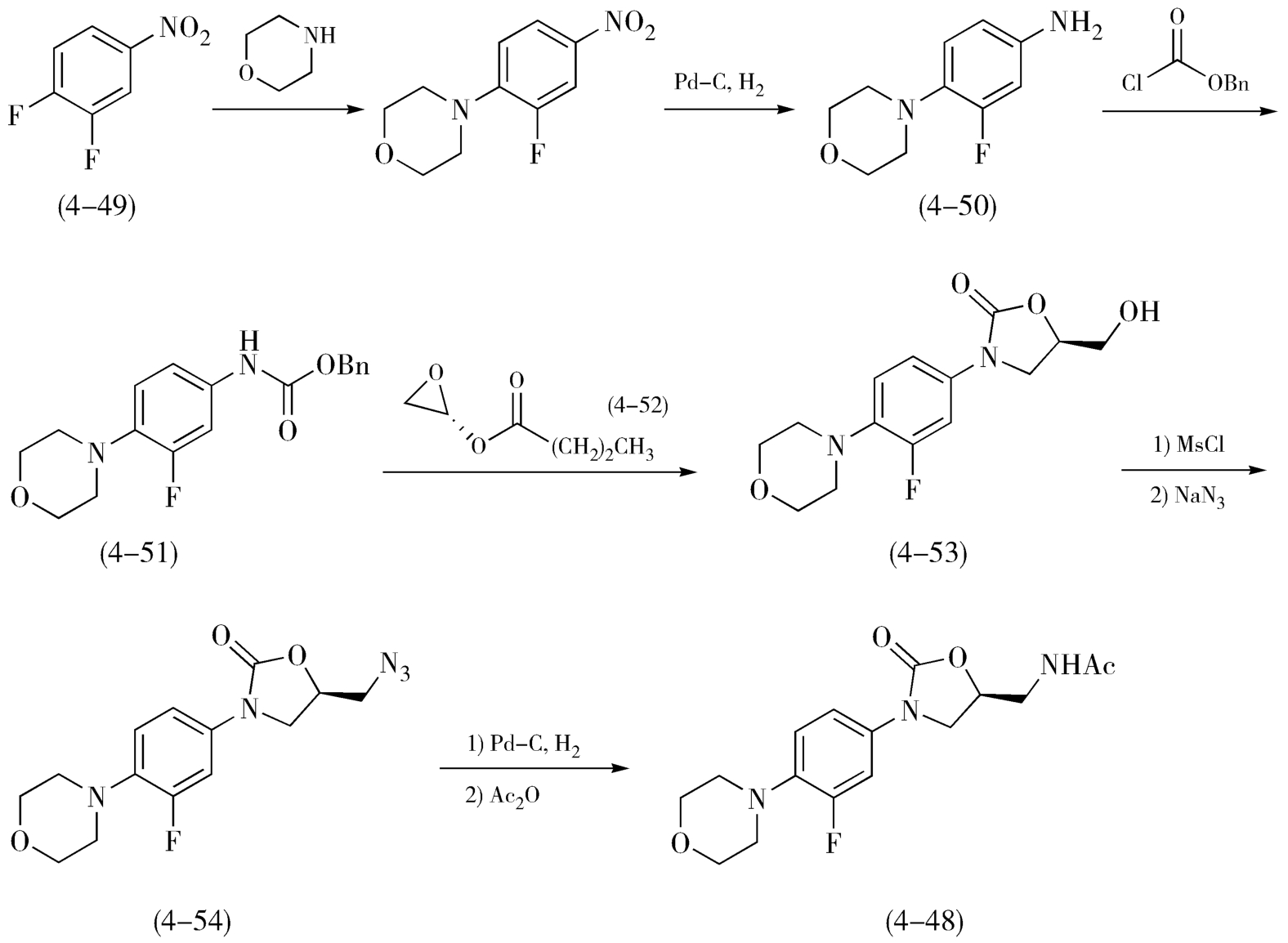
图4-21 利奈唑胺“直线式”合成路线
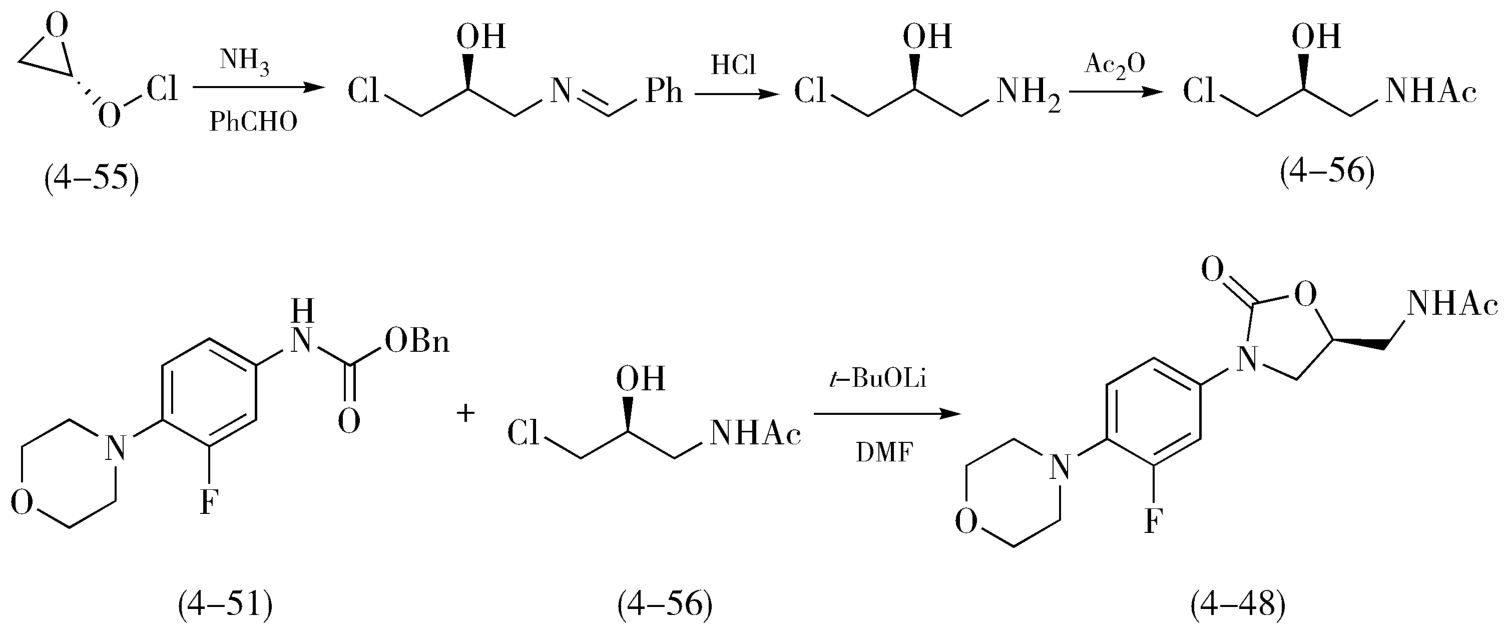
图4-22 利奈唑胺“汇聚式”合成路线
原辅料品种少、安全低毒、价廉易得、供应稳定。选择工艺路线首先应考察每一条合成路线所用的各种原辅材料,涉及所用原辅料的安全属性、价格成本、供应情况、规格和运输存储问题,其基本要求是利用率高、安全、价廉、有稳定的供应。尽可能选择无毒、无害或低毒、低害的试剂,避免易燃易爆、毒性较大、不易控制的试剂,如克霉唑合成路线(1)中用到了易燃易爆的格氏试剂,路线(2)中用到了有毒且难控制的氯气,成为了工艺路线的限制性问题(图4-23)。
《化学药物原料药制备和结构确认研究技术指导原则》指出:起始原料应质量稳定、可控,应有来源、标准和供应商的检验报告,必要时应根据制备工艺的要求建立内控标准。没有稳定的原辅材料供应,就不能正常组织生产。例如,在甲氧苄啶合成路线中,用没食子酸为原料的路线(图4-3)技术非常成熟,最后因为原料价格问题被迫放弃。因此在对设计的化学药品合成路线进行评价时,应将所采用的原辅料来源进行全面系统的考虑,不仅要衡量不同反应路线所需原料的单耗、成本、供应商水平,还应考虑原料的品质,如原料中含的杂质含量、种类及其对最终产品的影响;要对原辅材料的来源做充分的调研,原料的供应商、价格、产量、规格等。
反应条件温和、易于控制;优选“平顶型”。良好的路线应具备各步反应的工艺条件稳定、可靠,各步骤的反应条件比较温和、易于实现、易于控制、安全无毒;尽量避免高温、高压,或超低温等极端条件;尽量避免无水、无氧反应的苛刻操作。所谓的工艺条件是指影响反应的控制型因素,通常包括物料纯度、加料量、加料方式、溶剂及试剂含水量、催化剂、反应温度、反应时间、反应体系的pH值、搅拌条件等。例如,克霉唑的工艺路线,如图4-23,路线(1)以邻氯苯乙酸乙酯(4-57)为起始原料,在无水乙醇中先与苯基格氏试剂发生Grignard反应,得到叔醇(4-58),再经二氯亚砜氯代,制备了苄位上连有两个苯基的苄氯(4-59),最后在碱存在下与咪唑发生亲核取代反应,完成克霉唑(4-60)的合成,此法仅用了三步就完成了克霉唑的制备,原料易得,氯代和 N -烷基化反应收率都较高,产物质量好;但不足之处在于,使用了格氏试剂,整个反应体系要求严格无水,原辅料含水量和质量要求高,反应条件苛刻,使该路线的实际应用受到限制。路线(2)采用甲基氯苯(4-61)为原料,通过与氯气的氯代反应合成林氯苯三氯甲烷(4-62),再与苯通过F-C反应得到相应的苄氯(4-59),最后与咪唑反应制得克霉唑(4-60),这种方法合成路线简短、原辅料来源方便,但采用氯气进行氯化步骤中,一步要引入三个氯原子,反应温度较高、反应时间长,而且有大量反应的氯气容易溢出,后处理难以吸收完全,操控难度大,带来的环境污染、设备腐蚀和人员伤害较大。因此路线(1)(2)反应条件不温和、操控困难大、试剂安全性差、后处理问题多。路线(3)改用邻苯率甲酸(4-63)为起始原料,先与二氯亚砜反应制得邻氯苯甲酰(4-64),再与苯在三氯化铝催化性,进行Friedel-Crafts酰基化反应得到二苯甲酮类化合物(4-65),后者与氯化磷反应,形成苄位二氯代化合物(4-66),化合物(4-66)再发生一次与苯的F-C反应,得到的化合物(4-59),再经氨化得克霉唑(4-60),此路线收率也较好,明显较路线(1)(2)反应步骤多,但因为分两次引入苯基,每一步反应都条件温和、易于控制、成本较低,避免了上述氯气氯化、格式反应的苛刻条件和危险性,对环境的影响相对较小,该路线更适合与工业化生产。
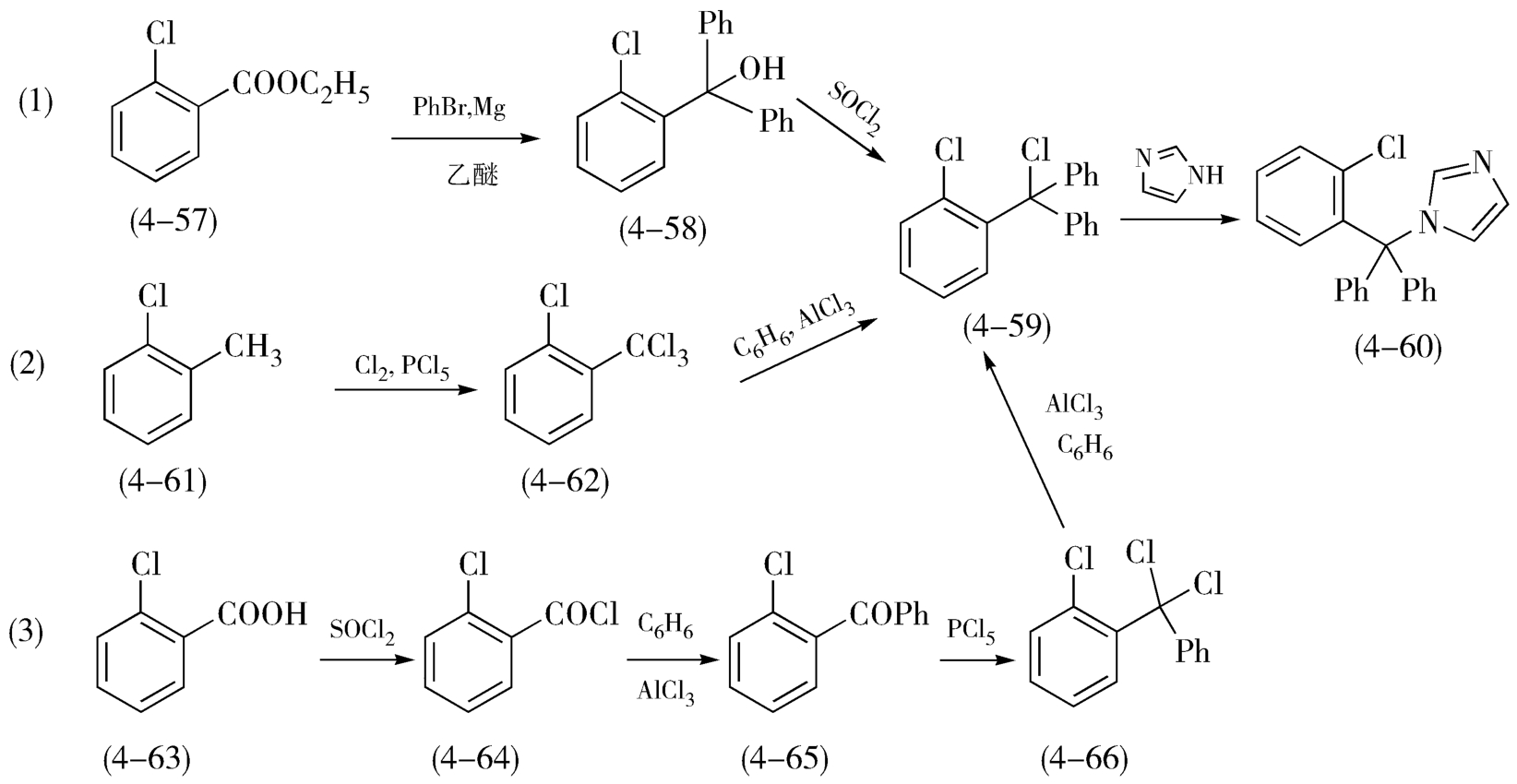
图4-23 克霉唑的合成路线
工业化价值较高的工艺条件,应在较宽的操作范围内实施,容易实现预期的反应过程和转化结果,得到理想质量和收率的产品,这样的反应,最佳反应条件范围较宽,即使某个工艺参数稍稍偏离最佳条件,其收率和质量也不会受到太大的影响,反应的结果与反应条件的关系所呈现的曲线为“平顶型”,我们称这种特点的反应为“平顶型反应”,如图4-24。与之相反,最佳反应条件和结果的关系曲线呈“尖顶型”,意味着如果工艺参数稍有变化,就会导致收率、质量明显下降,则属于“尖顶型反应”。“平顶型反应”条件稳定、副反应少、反应条件不苛刻、易于控制、整体风险小、生产操作灵活度高、劳动强度小;尖顶型反应,副反应多、反应条件苛刻、条件不易控,同时还关系到安全生产技术、“三废”防治、设备条件等,所以工业优选“平顶型”工艺条件。当然,对于“尖顶型”反应,若能达到更优越的结果,且在工业生产上可以通过精密仪器、自动控制等手段予以实现的,也可为现代化制药、智能制药工业生产所采用。
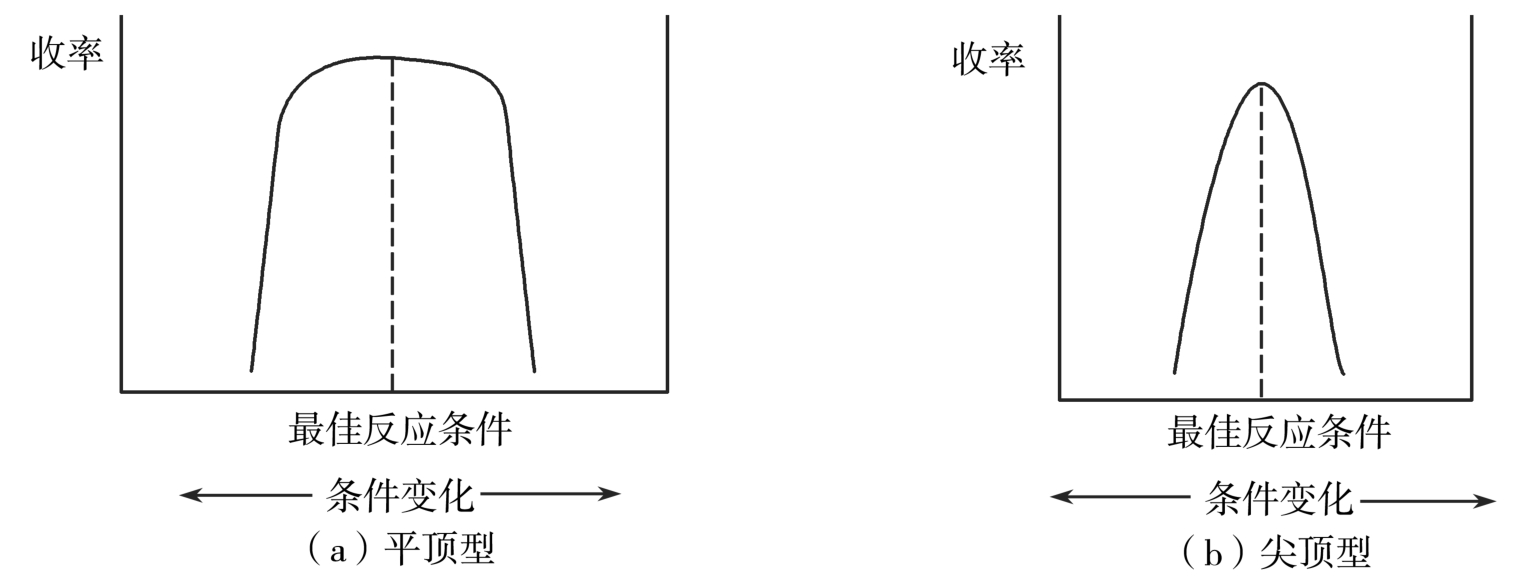
图4-24 “平顶型”反应和“尖顶型”反应示意图
例如,在芳环上引入醛基的过程可以采用多种途径实现,如图4-25,其中的途径(5)的反应需要20小时以上,副反应多、收率低、产品又易聚合,常生成大量树枝状物,从而增加后处理的难度,属于“尖顶型”反应。途径(6)为Daff反应,是在活泼芳环上引入甲酰基的经典反应,所用甲酰化试剂为六次亚甲基四胺(即乌洛托品),反应条件易于控制、操作简便、产物易纯化,是典型的“平顶型”反应。途径(2)是Gattermann-Koch反应,也是经典的芳环甲酰化反应,但该反应以毒性较大的CO和HCl为原料,需要使用加压设备,反应条件控制难度较大,属于典型的“尖顶型”反应,但是原料价格低廉、收率很好,且实现了生产过程的自动控制,目前已为工业生产所采用。
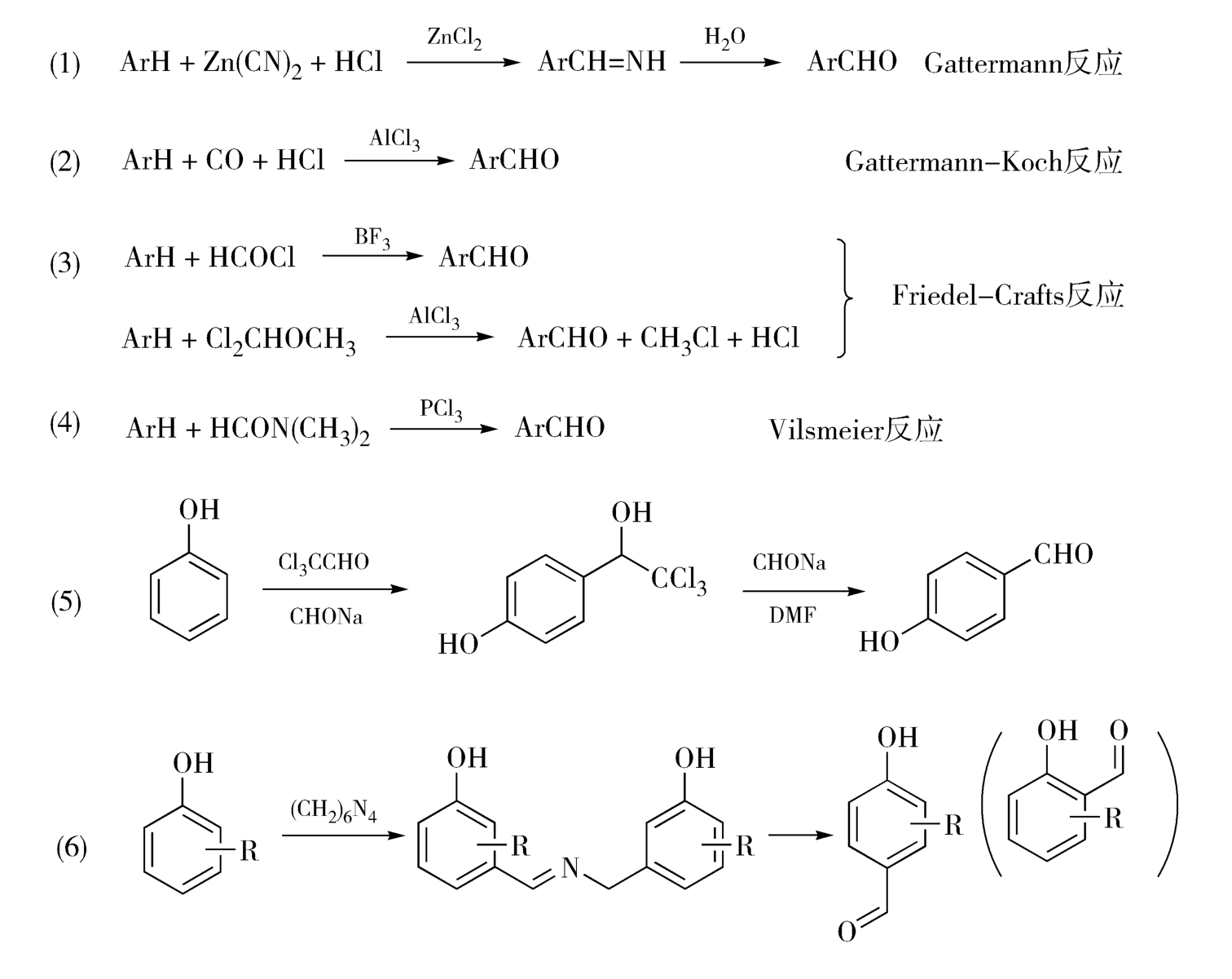
图4-25 芳环甲酰化反应示意图
副反应少,中间体和产物易于分离、纯化;优选“一勺烩”。分离和纯化是工艺路线设计的重要组成部分。在药物合成路线中,副反应少往往意味着该步反应收率较高、杂质少、后处理简单、产品易于分离纯化,这样能很大程度简化工艺、节约成本,最终产品的质量易于达到标准。
例如,酰胺键的构建是药物合成中最常见的反应,可以通过很多方法实现:①羧酸与胺的缩合酰化反应。②酰卤与胺的酰化反应。③氨与酸酐的酰化反应。④氨酯交换反应。⑤氰基的水解。⑥Overman重排、Schmidt重排、Beckman重排等重排反应,其中应用最广泛的是前两种,但第一种方法,羧酸与胺的缩合反应,反应条件易行,收率稳定,可采用多种缩合剂,适用范围广,比较灵活,但因为引入了缩合剂,反应完毕后反应液中含有复杂的缩合剂消除物,这些离去基组成的新物质及残留的缩合剂难于除去,需经柱层析纯化,后处理相对麻烦、溶剂消耗大、处理周期长、风险大,所以往往选择第二种途径,酰卤与胺的酰化反应,经萃取、浓缩即可分离、纯化,易于后处理,更利于工业生产。
理想的工艺路线应尽可能具备副反应少的特点,甚至在某些情况下中间体无需纯化就进入下一步反应,俗称“一勺烩”。所谓“一勺烩”,是指某步化学反应完毕后,反应液体系里的试剂、副产品、溶剂等对下一步反应影响不大时,可先不做分离、纯化,直接进行下一步,将几个反应连续操作,实现多步反应后处理的“一锅操作”。“一勺烩”使用得当,能最大程度的简化流程,节约时间,减少成本,降低生产劳动强度。
例如,在抗菌药甲氧苄啶(Trimethoprim,TMP,4-67)的合成过程中,如图4-26,丙烯腈(4-68)在甲醇钠催化下与甲醛加成生成三甲氧基丙腈(4-69),反应液不需处理,可直接加入中间体3,4,5-三甲氧基苯甲醛(4-70),在甲醇钠催化下缩合生成单甲醚(4-71),单甲醚(4-71)继续在甲醇钠催化下发生双键重排得到二甲醚(4-72),这3步反应催化剂相同、溶剂相同,故而采用“一勺烩”的方式连续进行,最后,所得二甲醚(4-72)与硝酸胍反应制得甲氧苄啶(4-67)。
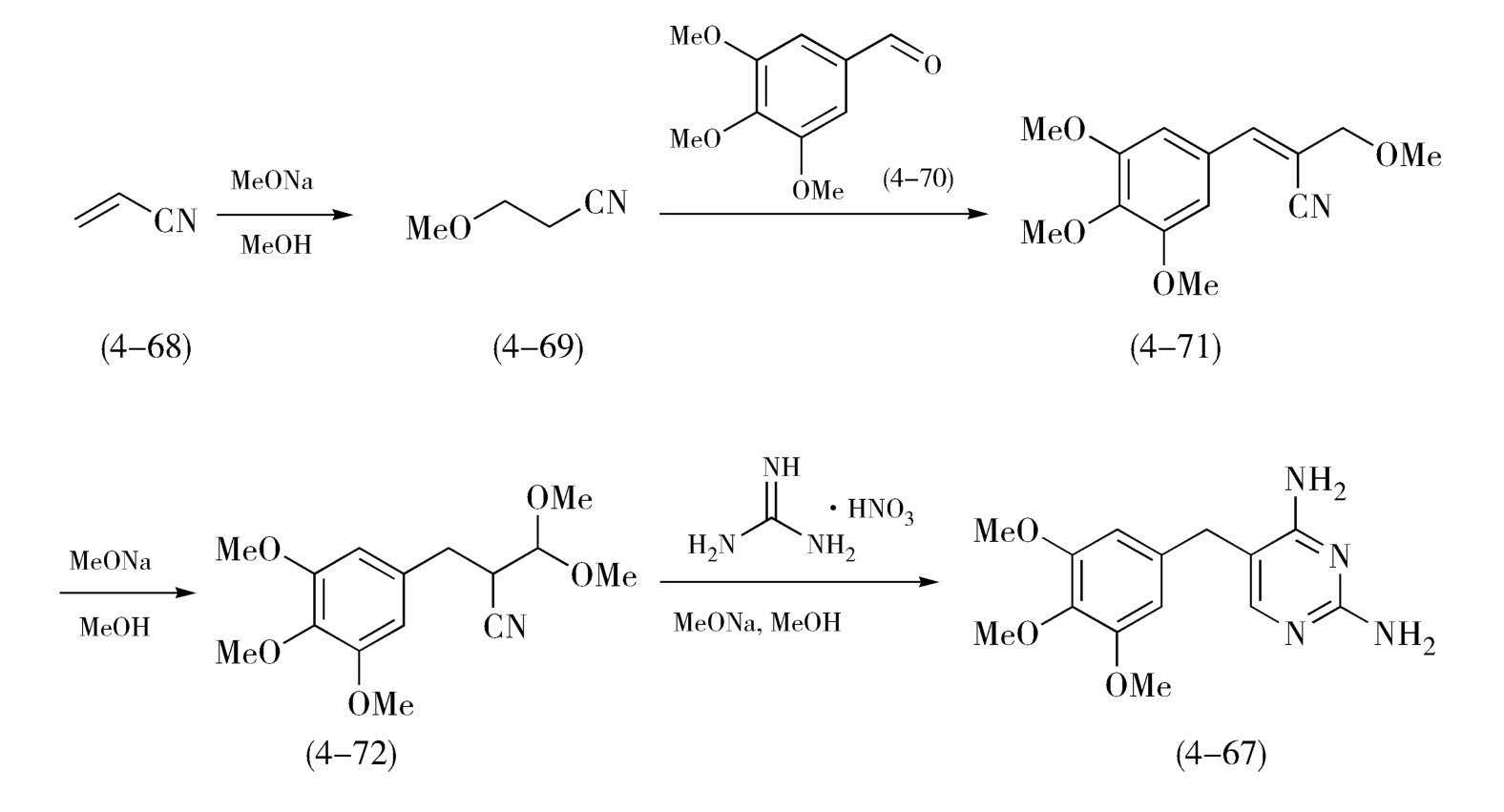
图4-26 甲氧苄啶的合成路线
再如,吡罗昔康(Piroxicam,4-73)的合成工艺也是“一勺烩”的典型案例,从原料邻苯二甲酸酐(4-74)出发,共需经历13步化学反应,该路线为“直线式”合成法,且步骤较多,但由于采用了几步“一勺烩”工艺而显现出独特的优点。如图4-27,由原料邻苯二甲酸酐(4-74)出发,经(1)氨解、(2)Hofman降解、(3)酯化3步反应制得邻氨基苯甲酸甲酯(4-75),这3步反应的副产物较少,几乎不影响主产物的生成,且3个反应都在碱性甲醇溶液中进行,故可以连续进行,合并为第一个工序。邻氨基苯甲酸甲酯(4-75)经(4)重氮化、(5)置换和(6)氯化3步反应生成了2-氯磺酰基苯甲酸甲酯(4-76),此3步反应均需在低温酸性液中进行,生成的氯代物(4-76)转入甲苯溶液中得以分离,此3步反应可连续操作,合并为第二个工序。(7)胺化、(8)析酸两步反应可以连续进行,为第三个工序,其产物为糖精(4-77)。糖精(4-77)经(9)成盐反应得糖精钠(4-78),再经(10) N -烷基化、(11)碱催化重排扩环和(12) N -甲基化反应得到苯并噻嗪酯类中间体(4-79),这几步反应都在碱性条件下进行,无需分离中间体,可直接进行连续操作,成为第四个工序。最后噻嗪酯类中间体(4-79)与 α -氨基吡啶发生(13)酯的氨解反应形成酰胺键,完成吡罗昔康(4-73)的合成。在上述路线中,四次巧妙地使用“一勺烩”,不仅明显地减少了后处理的次数,简化后处理的过程,而且显著地提高了整个路线的总收率,使吡罗昔康的生产成本大幅降低,是典型的“一勺烩”工艺路线。需要注意的是,减少后处理的次数和简化后处理的过程是有一定风险的,过分地采用“一勺烩”工艺,可能会导致产物或重要中间体纯度的下降,使分离纯化的难度增加,甚至可能影响产品的质量。
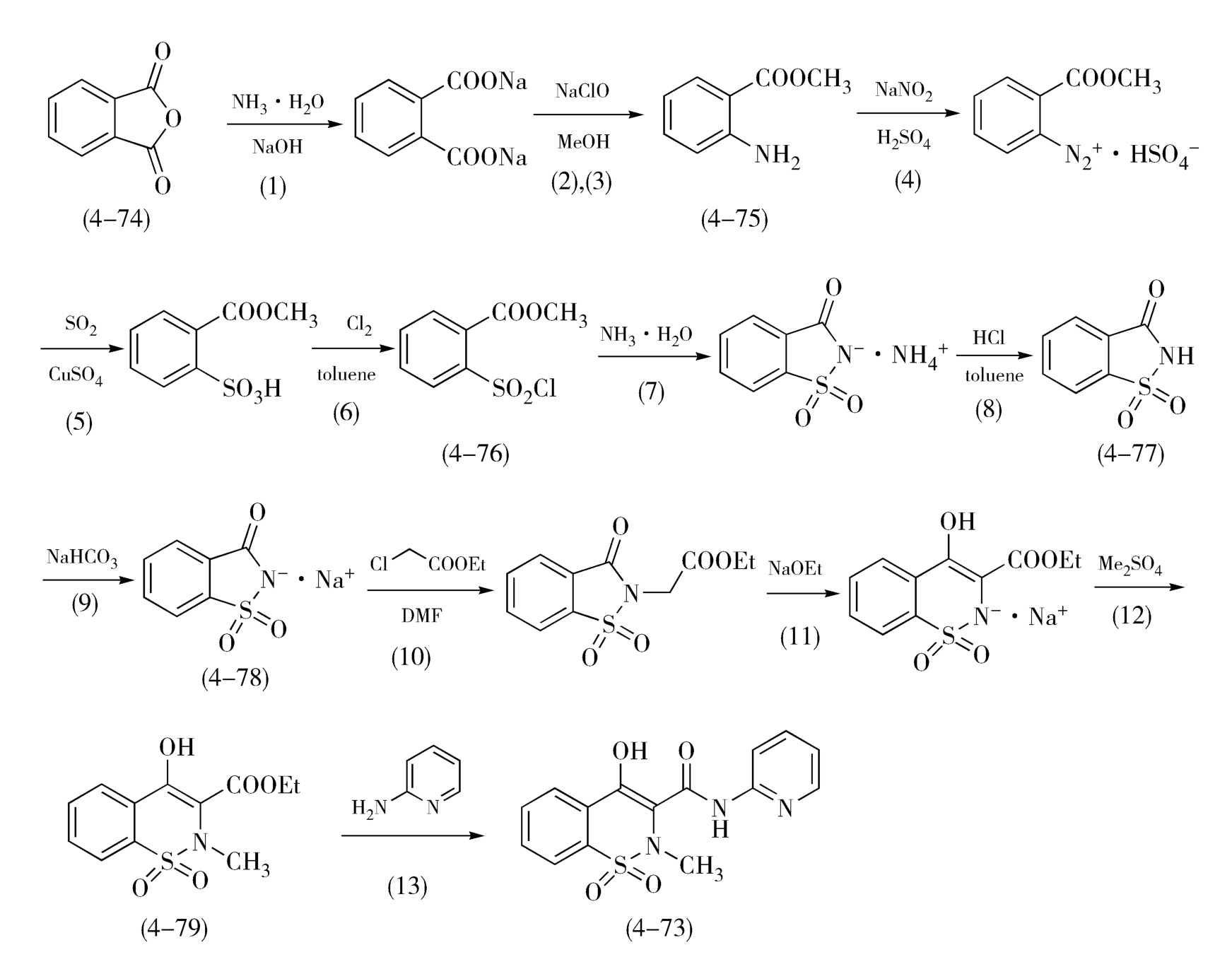
图4-27 吡罗昔康的合成路线
当然,不是所有的工艺路线都可使用“一勺烩”,若使用不当,也可能存在风险:造成副产物增多、分离纯化难度增加、目标产物纯度下降、产品质量达不到药用标准等。
“三废”少且易处理;“原子经济性”好。传统的药物合成化学方法及相关的制药工业给人类带来了巨大的贡献,然而由于化学制药需要使用大量的化学品,并可能产生大量的废水、废气、废渣,也对人类赖以生存的生态环境造成了严重污染。现代化的制药工艺秉承可持续发展的宗旨,提出了“绿色化学”理念,核心是“原子经济性”理论。“绿色化学”是指在化学品生产时应有效利用原料,消除废物,避免使用有毒和危险的化学试剂或溶剂。
“原子经济性”是指在合成路线中,参与化学反应的试剂应尽可能多地转化到最终产物中去,实现原子的高效利用。理想的原子经济性反应,是原料分子中的原子百分之百转变到产物中,实现废物的零排放。原子经济性可用原子利用率这个指标来表示,所谓原子利用率,直接反应化学结构中原子被利用的程度,原子利用率的计算,如公式4-1所示,用产物的分子量除以所有参与反应的反应物的分子量之和(注:因为催化剂在反应前后其质量原则上不发生变化,不计入加和)。

公式4-1 原子利用率计算公式
以布洛芬的合成为例,如图4-28、图4-29所示,Boots法的原子利用率为40%,原子经济性不好,意味着有很多原子以废物的形式排出,需要进行大量的“三废”处理;而BHC路线的原子利用率达到了77.4%,如果再考虑到副产物醋酸在生产中被回收再利用,那该路线原子利用率可趋近100%,意味着有很少的废物产生。BHC路线正是因为原子利用率高而实现了“三废”少、后处理简单、副产物与催化剂可回收利用、总收率高的的特点,是制药工业史上原子经济性极高的经典路线,切实实现了环境影响的最小化,因此获得了1997年美国“总统绿色化学挑战奖”。前面提到的西他列汀的改进路线同样也是原子经济性的典型案例。
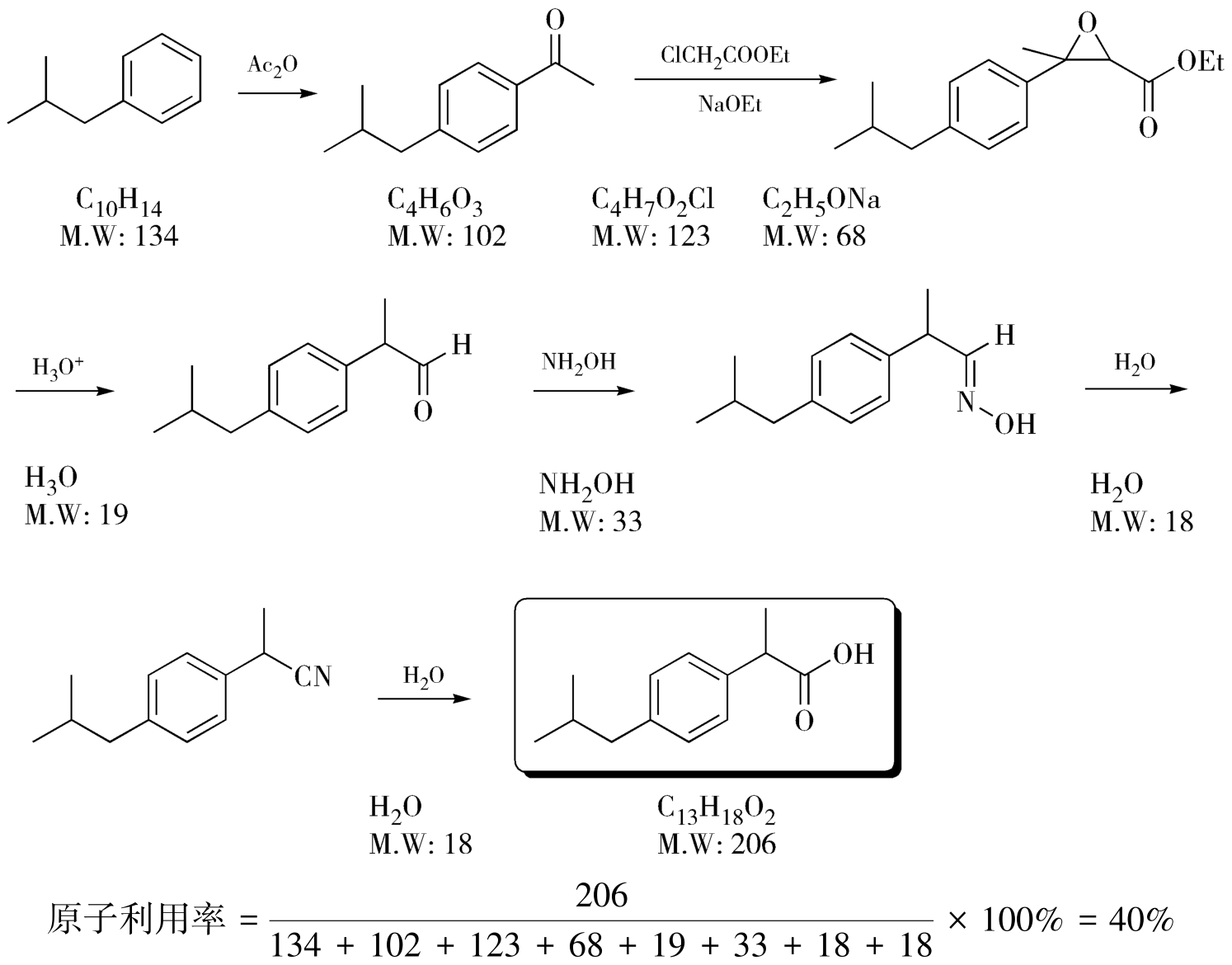
图4-28 布洛芬Boots法合成路线的原子利用率
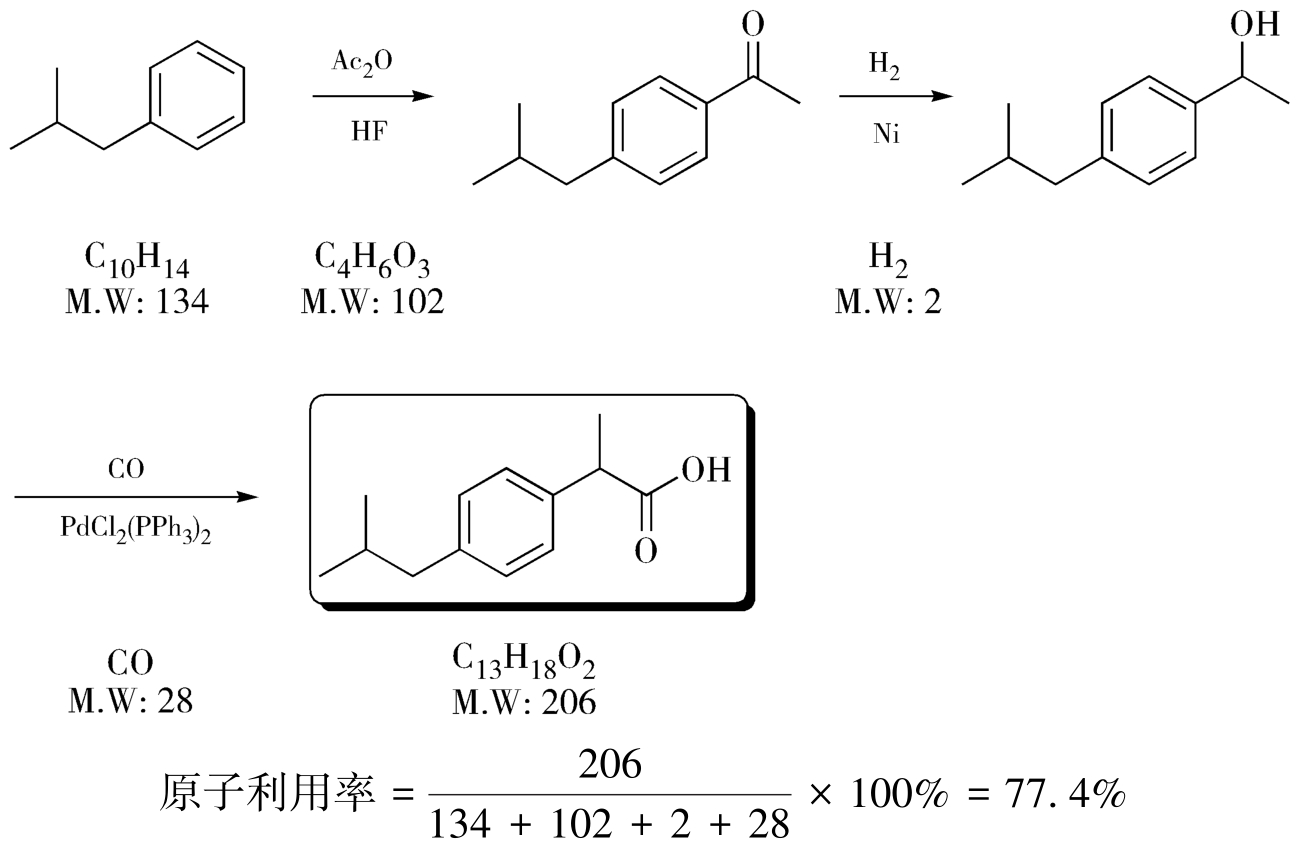
图4-29 布洛芬BHC法合成路线的原子利用率
设备条件要求不苛刻;推进现代化制药设备的使用。
在工业化合成路线选择时,必须考虑设备因素。生产设备可靠性是评价合成工艺路线的重要指标。实用的工艺路线应尽量使用常规设备,最大限度避免使用特殊种类、特殊材质、特殊型号的设备。大多数光化学、电化学、超声、微波、高温或低温、剧烈放热、快速淬灭、精准控温、高度无水、超强酸碱、超高压力等条件,需要借助于特殊设备来完成,在实验领域均可以实现,但是出于技术、规格、成本、安全性等因素的限制,在工业化生产中,应尽可能避免。例如,微波加热技术已经是实验室的常规技术手段,多种型号的微波反应装置可供选择,然而,可用于工业化的大型微波反应器,目前尚未上市。又如,对于使用丁基锂、二异丙基氨基锂等有机金属化合物的反应,通常要在极低温度下进行,在实验室用干冰丙酮浴比较容易实现,但在实际生产过程中,不仅要有大功率制冷设备,还要求设备具有优良的保温性能,达到精准控温,否则反应体系的温度由于难以均衡而产生大相径庭的结果。再例如加压反应,即便是高压反应,虽然在化学工业生产中已大规模普遍应用,但毕竟高压反应器对密封、承压能力、设备材质等要求高,而且隐患风险相对较大、安全措施耗费大、涉及面广、成本较大,所以,路线选择和改造时能避免最好。
随着科学技术不断进步,设备与材料的生产限制已越来越小,因此对于能够显著提高收率,可实现机械化、自动化、连续化,显著提高生产效率、利于劳动保护及环境保护的工艺路线,即使设备要求高些,也应根据现有条件尽可能予以考虑,这是体现工艺路线先进性的关键,是现代化制药、智能制药的重要组成。例如,制药工艺自控系统已在各大制药企业广泛应用,DCS控制技术带来了制药设备的系统化改进,是智能制药模式的巨大突破,因此,在制药工艺改进中,应努力推进智能化设备的使用。
针对手性药物,充分开发不对成催化合成及手性拆分工艺。手性药物占据全球药品市场的重要地位,在现有临床药物中占60%以上。手性药物具有疗效好、毒副作用小、技术门槛高、经济效益好的特点。现代药物研发的一个核心焦点就是如何高效得到一定手性的终产物,目前手性药物的制备主要有两大途径:手性拆分和不对成合成。拆分法对外消旋的手性药物或其手性中间体进行分离,主要包括结晶拆分法、化学拆分法、色谱拆分法、生物酶拆分法等;不对称合成,又称手性合成,指在手性试剂、催化剂或助剂等手性底物的作用下合成得到所需光学特征的目标手性分子。手性药物合成路线的选择和改造是高效合成的根源,是手性药物研发的核心任务、热点方向,也是其高效工业化的期待。
金属有机催化剂在不对称手性合成中的应用,带来了新合成方法的产生,使手性药物合成途径大幅简化,实现高效合成、节能增效,这也是绿色化学消除污染、保护环境的核心着手点。例如,默克公司对西他列汀(Sitagliptin,4-80)工艺路线的改进,就因应用了新型催化剂和更换原料,优化了之前的路线,合成效率发生了巨大的突破。西他列汀为Ⅱ型糖尿病治疗药物,最初的合成生产路线需8步反应,见图4-30,Schollkopf手性试剂(2 S )-2,5-二氢-3,6-二甲氧基-2-异丙基吡嗪(4-81)与2,4,5-三氟溴苄反应得化合物(4-82),化合物(4-82)与盐酸盐反应开环后用(Boc) 2 O保护氨基得化合物(4-83),酯基水解得对应的酸(4-84),再与氯甲酸异丁酯和重氮甲烷反应得重氮羰基化合物(4-85),经Wolf重氮酮重排得烯酮后与甲醇发生Arndt-Fistert反应得酯(4-86),水解酯基得相应的酸(4-87),酸(4-87)与杂环化合物(4-88)经缩合、脱保护得西他列汀(4-80),其中用到缩合剂等几个分子量较高的试剂,这些试剂中的大部分原子并没有转移到最终产物中去,只能以废料形式排出,所以整体原子利用率不高,路线中又涉及氨基的保护和脱保护,步数较多。经摸索发现,如图4-31,不经保护的烯胺(4-89)不对称加氢也可以实现最终转化,因此应用了二茂铁配体铑催化剂不对称催化合成得到了高光学纯度、高产率的西他列汀,也开发出一条以手性原料 β -氨基酸(4-90)为原料,直接提供目标分子中手性片段的高效合成路线。新路线可高效得到手性目标产物,且省去了基团保护步骤,可实现3步转化,同时原子利用率高,每生产1kg的西他列汀可减少220kg废物的产生,且总产率提高了近50%,原材料消耗量、合成时间、能量和废料的产生量亦大幅减少,成本更节约,有利于环境保护。
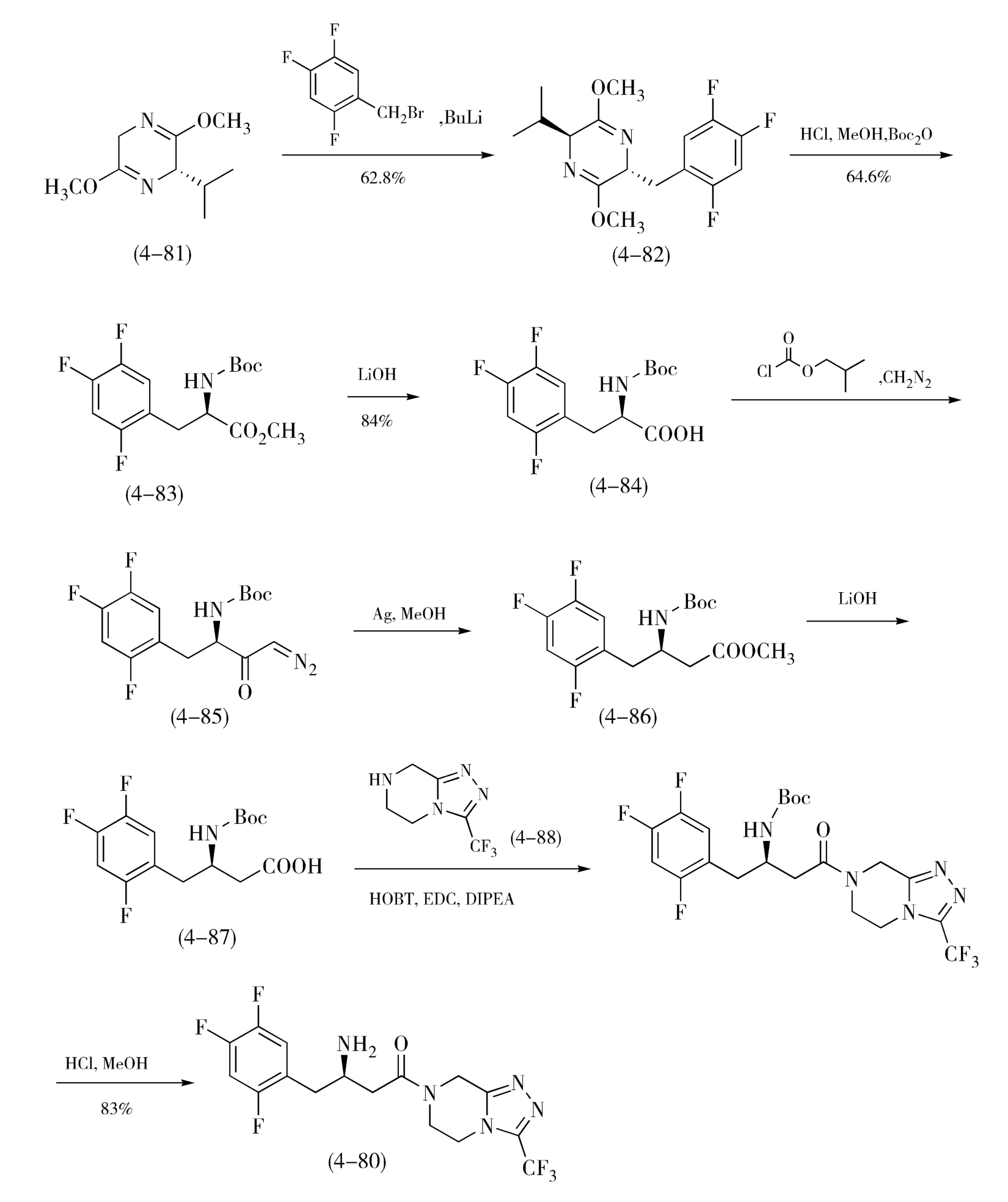
图4-30 西他列汀的“8步法”合成的路线
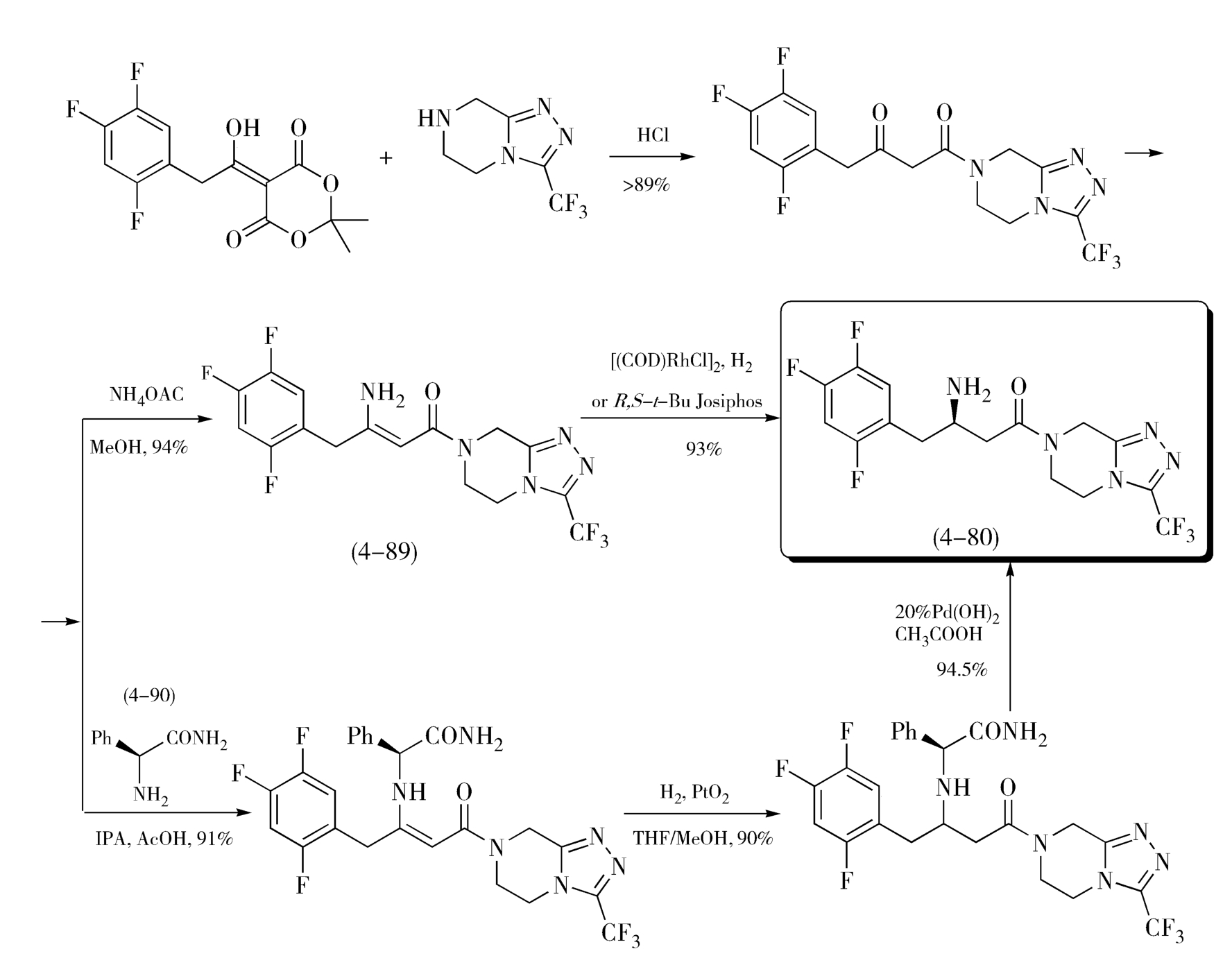
图4-31 西他列汀的优化合成路线
关于手性拆分,依靠对产物消旋体进行手性分离是很难实现工业化的,若能利用溶解性、成盐等物理特征差异便捷地先获取具有目标光学特征的片段,以其为原料或中间体,再进行手性药物的合成,那实属难能可贵,会最大程度地提高效率、简化流程,具有很高的工业化生产价值和潜能。例如,在氯霉素的合成中,就采用了结晶法拆分法,利用某中间体的外消旋体与L-酒石酸成盐形成的一组非对映异构体在甲醇中的溶解度差异较大进行了拆分;也可以采用单旋体晶种诱导、交叉往复进行的诱导结晶法,这是手性拆分成功工业化的经典案例。再如,瑞德西韦的合成工艺,也是合理利用中间体的手性异构体在溶解性上的显著差异,进行手性药物合成路线选择和改造的优秀新作。瑞德西韦(Remdesivir,4-90b),是针对新型冠状病毒(2019-nCoV)最早的、也是被寄予厚望潜在药物,现在处于临床阶段。瑞德西韦(Remdesivir)是美国吉利德·科学公司开发的一种核苷类似物,是一种RNA聚合酶(RdRp)抑制剂,其抗病毒的作用机制是通过抑制病毒核酸合成。瑞德西韦的研究与开发备受全世界关注和期待。图4-32是瑞德西韦的初始合成工艺,该路线使用内酯(4-91)和化合物(4-92)在正丁基锂或者氢化钠等强碱作用下发生糖苷化得到中间体(4-93);中间体(4-93)中的羟基被氰基取代后得到中间体(4-94);然后在三氯化硼作用下脱除苄基保护得到化合物(4-95);化合物(4-95)与化合物(4-96)缩合即可得到消旋产物(4-97)。得到消旋产物是该路线最大的不足,这要求在最后一步不得不采取手性HPLC来进行手性拆分得到光学纯的化合物瑞德西韦(4-90b),这显然不能满足原料药的大批量生产。因此,在初始合成工艺的基础上,吉利德又开发了瑞德西韦的二代合成工艺,如图4-33,新工艺中避免了使用丁基锂、氢化钠这样的危险试剂、优化了各步骤产率。最关键的是,在新的工艺中,研究人员发现中间体(4-98)的两种构型在异丙醚中具有不同的溶解性,由此可以快速大量获得手性纯的中间体(4-98b),这成功避免了终产物的手性拆分,为瑞德西韦(Remdesivir)的批量生产奠定了基础。
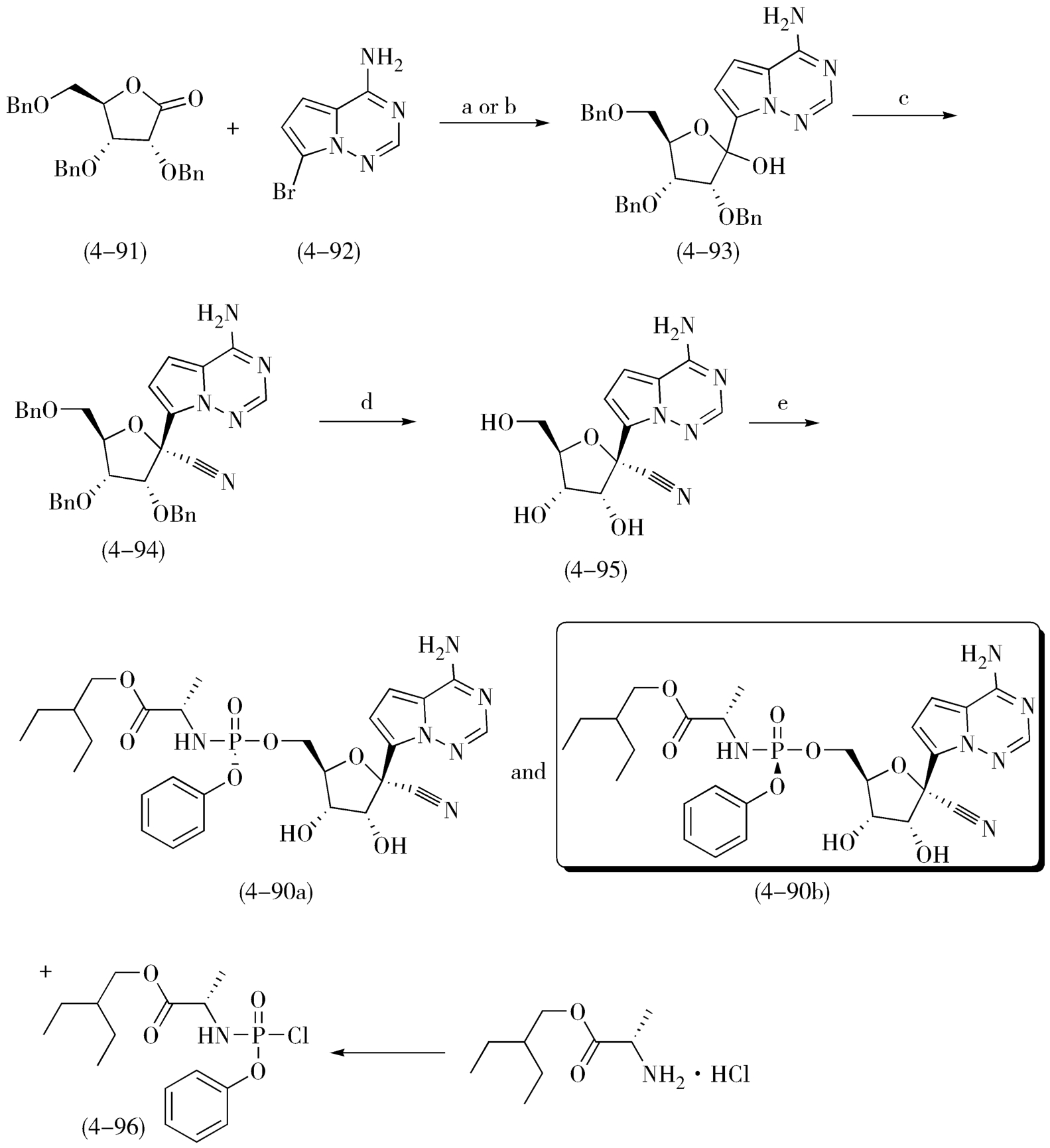
图4-32 瑞德西韦起初的合成路线
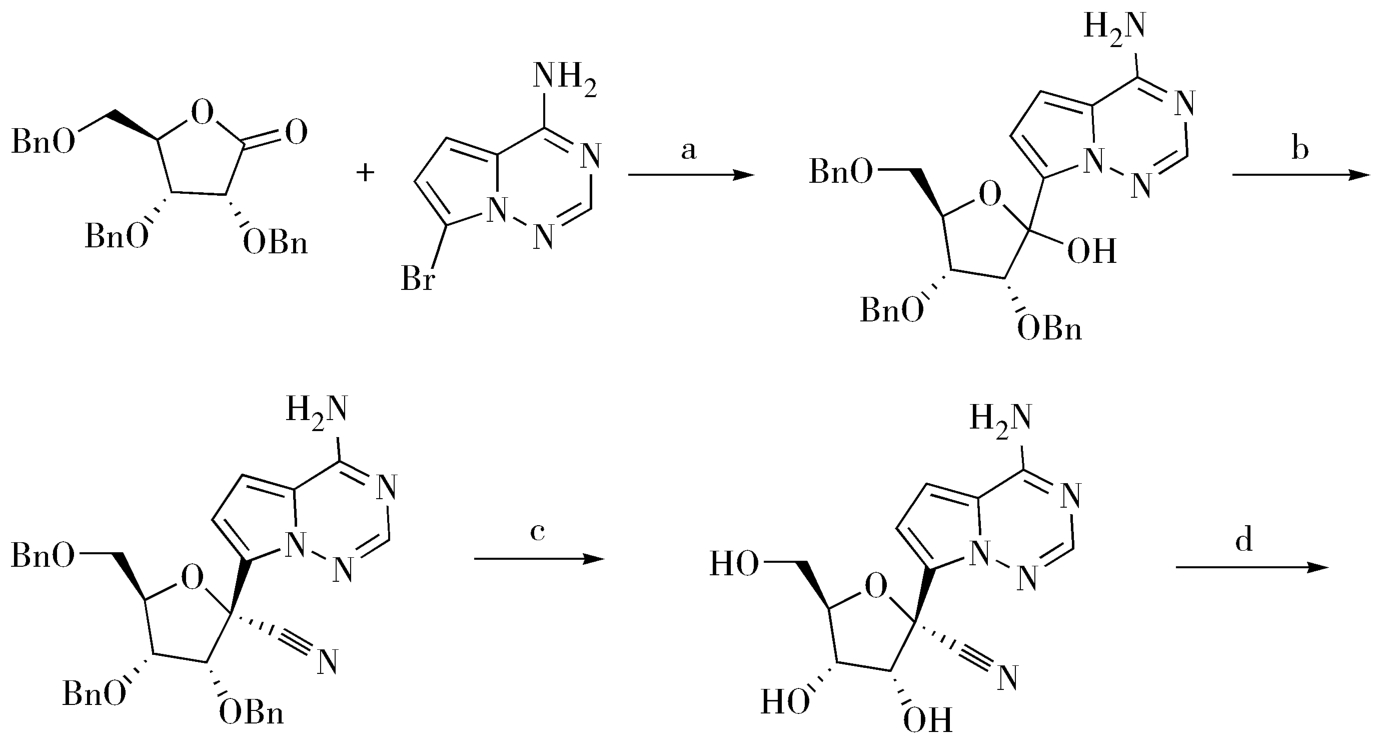
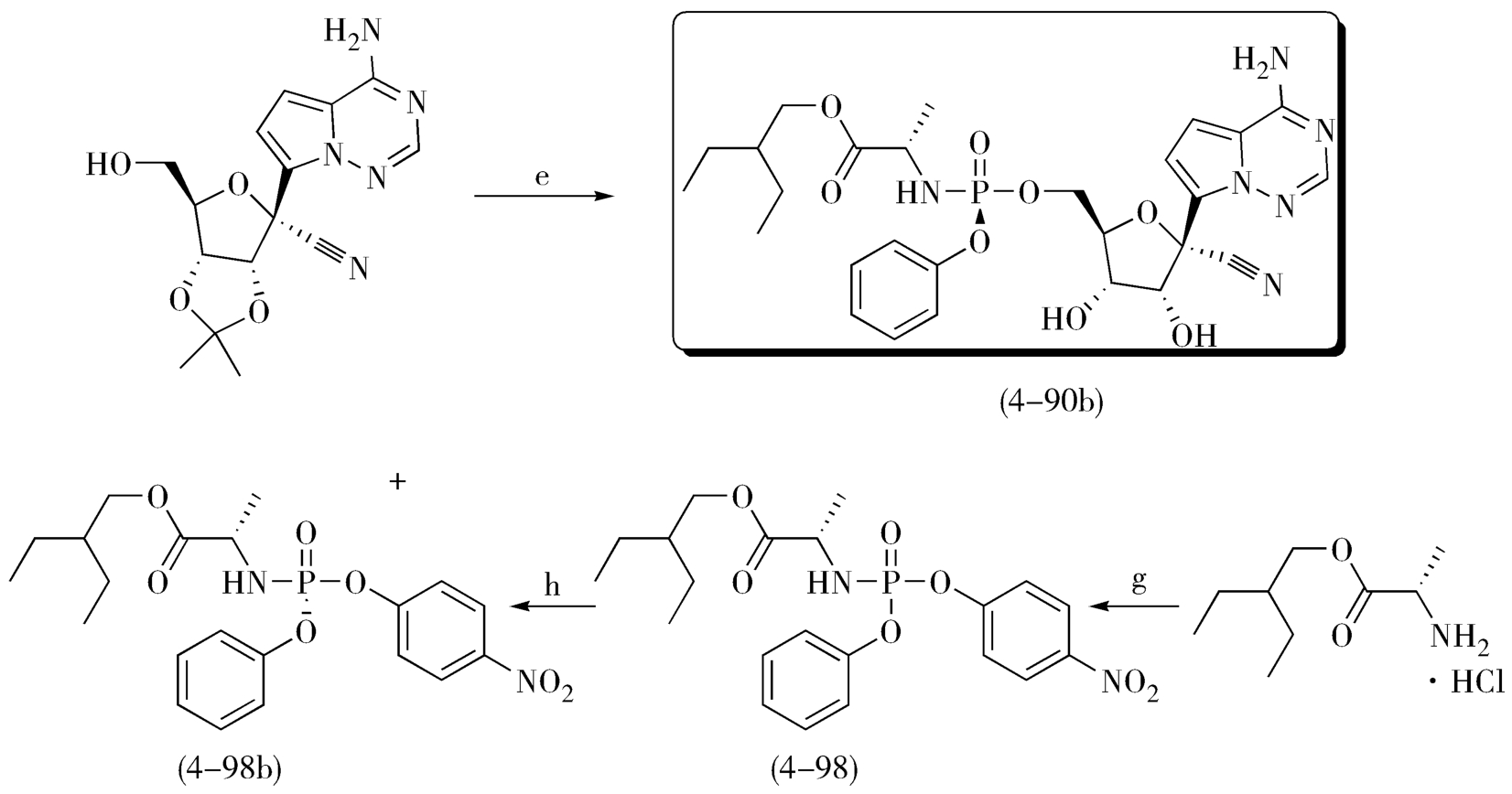
图4-33 瑞德西韦的优化合成路线
收率最佳、成本最低、经济效益最好。以上七个方面,是评价药物合成工艺路线的主要内容;是选择工艺路线的基本原则;也是改造现有工艺路线的基本切入点。并不是实际投入的生产路线都能具备以上特点,但却是我们在设计工艺路线时应重点考虑的。当不能全部满足所有要求时,或各路线各有特点时,常用综合结果来考量,即在安全第一的前提下,以第八条为最直观的总原则:收率最佳、成本最低、经济效益最好。