
下载掌阅APP,畅读海量书库
立即打开



逆合成分析方法是合成药物的最基本方法,但是通过对药物分子结构和性质的考察,我们能够发现许多更为便捷有效的合成途径,而没有必要对每一个药物都进行繁琐的逆合成分析。下面介绍几种常用的合成药物方法。
对某些药物或中间体进行结构剖析时,常可发现其中存在分子对称性(molecular symmetry),这对于合成路线的设计工作是非常有益处的。具有分子对称性的化合物往往可由两个相同的分子经化学合成反应制得,或在同一步反应中将分子的相同部分同时构建起来,这就是分子对称法。
1.利用对称性合成己烯雌酚 如己烯雌酚(2-2)可以通过两分子的对甲氧基苯丙酮(2-42)在钛催化的情况下发生McMurry反应生成关键中间体(2-43),再经水解反应制得(图2-18)。
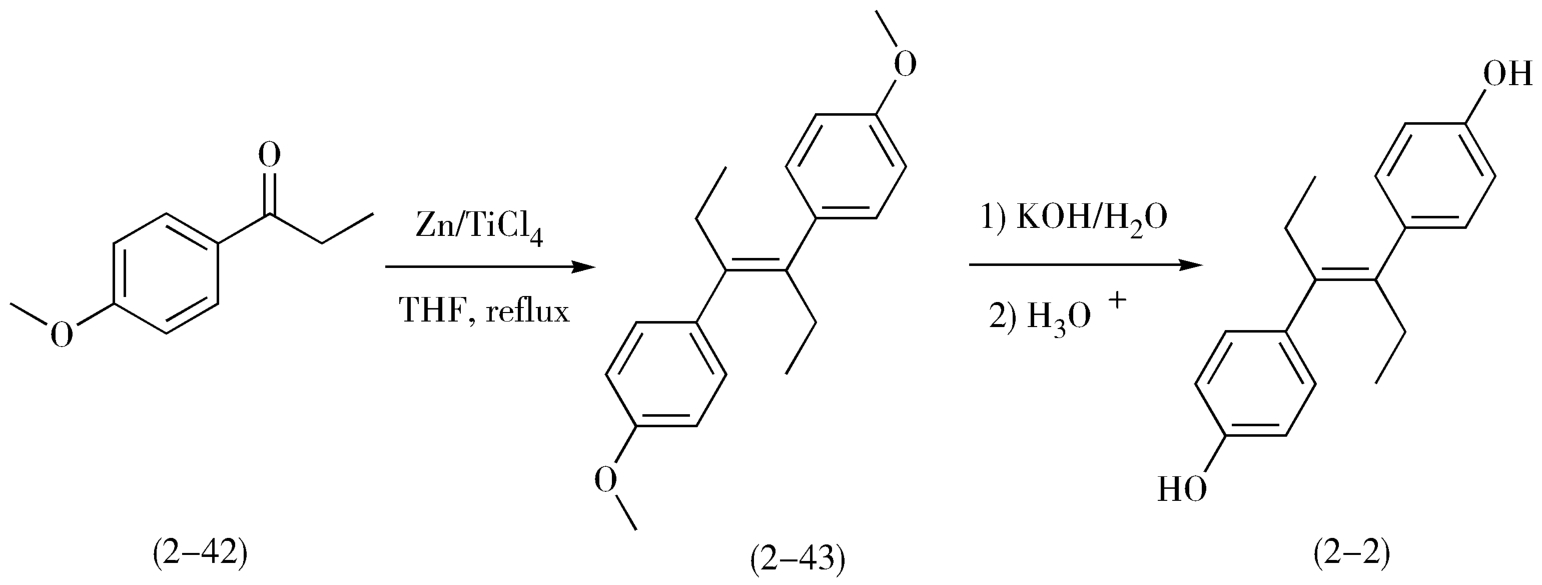
图2-18 己烯雌酚的合成
2.潜在对称性 目标药物分子的对称性,包括轴对称和面对称,都可用于简化合成设计,对于简单分子而言这一点是明显的。但有些药物分子本身并没有对称因素,只有经过一定的逆合成转化,才能得到一个对称的分子或一条对称的合成路线,从而简化合成设计,这叫作分子的潜在对称性(potentialmolecular symmetry)。例如,抗麻风药物氯法齐明(clofazimine,2-44)可看作吩嗪亚胺类化合物,即是2-对氯苯氨基-5-对氯苯基-3,5-二氢-3-亚胺基吩嗪(2-45)的衍生物,从2-45虚线处可看成两个对称分子(图2-19)。
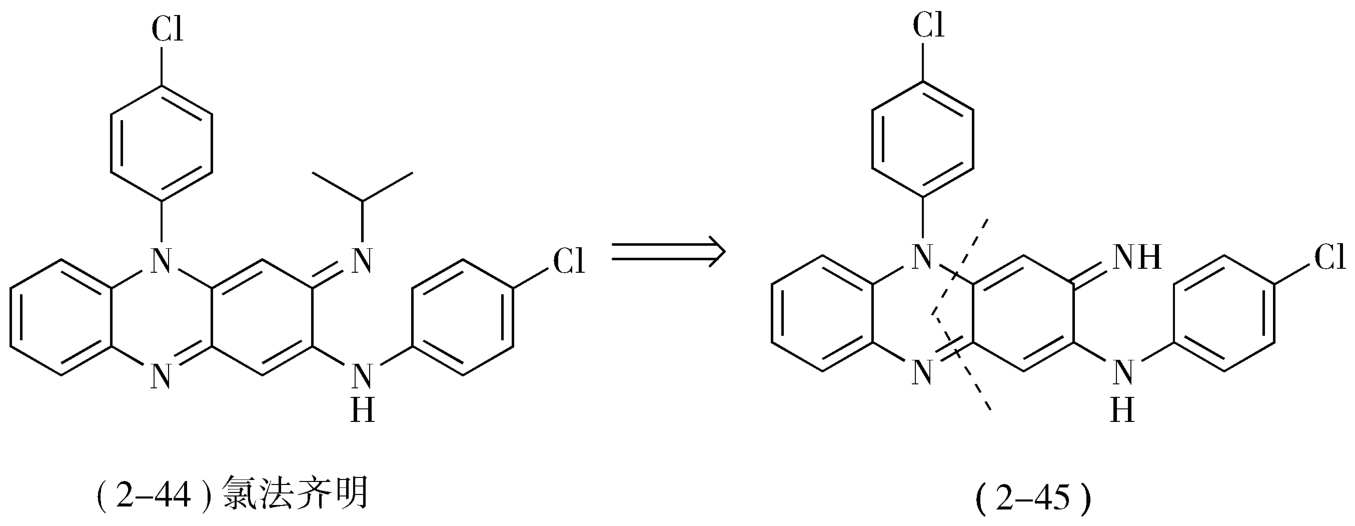
图2-19 氯法齐明逆合成分析
因此,可使用两分子的 N -对氯苯基邻苯二胺(2-46)在三氯化铁存在下进行缩合反应,然后与异丙胺进行加压反应,即可制得氯法齐明(2-44)(图2-20)。
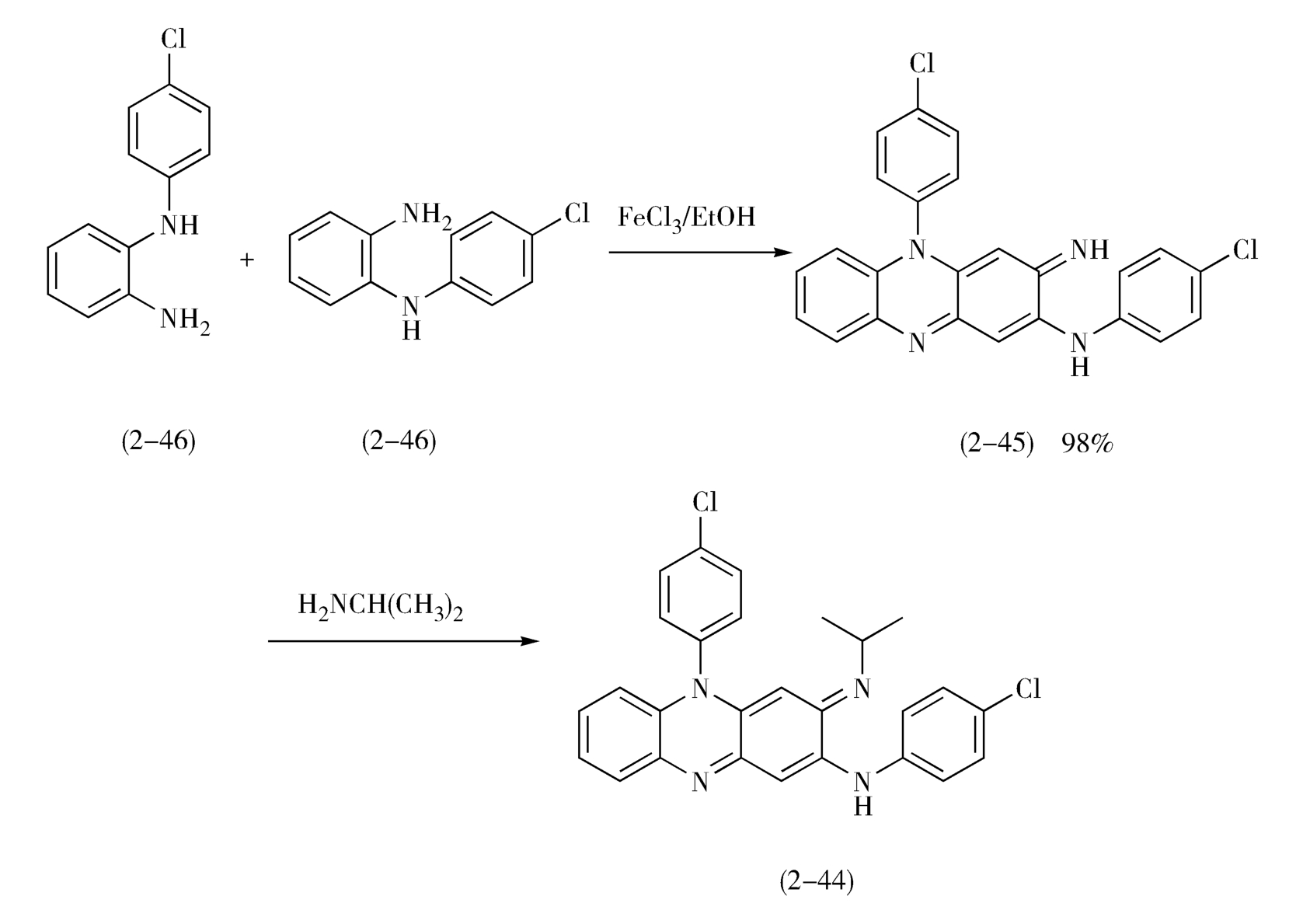
图2-20 氯法齐明的合成
除草剂地衣酸(usnic acid,2-47)本身也无对称性,但可分拆为相同的两部分,即可由相同两片段(2-48)汇聚合成,这时的合成是左右对称的(这种情况被称为自反性),下面的工作就是合成的2-48了(图2-21)。
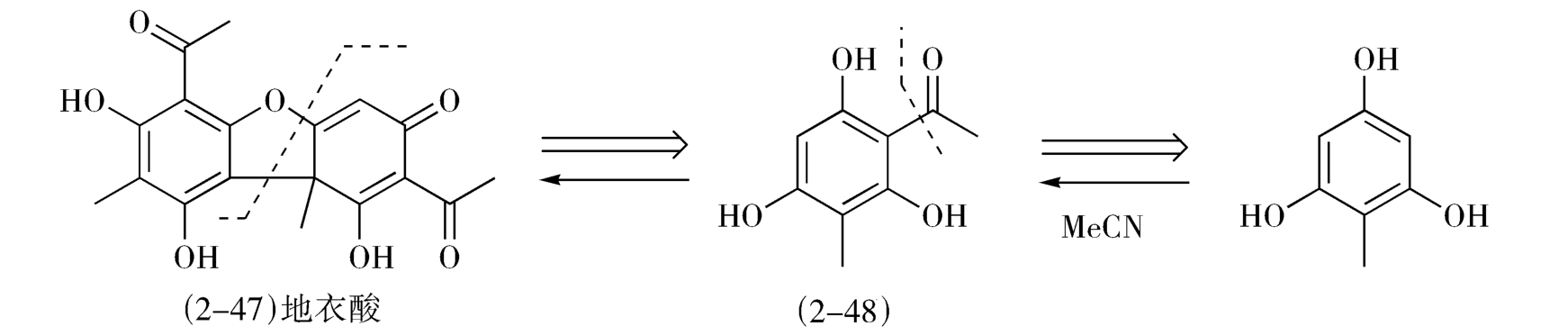
图2-21 地衣酸逆合成分析
对于一些药物或关键中间体,可根据它们的化学结构类型和官能团性质采用类型反应法进行药物工艺路线的设计。类型反应法是指利用常见的典型合成反应和合成方法进行路线设计的方法。类型反应法在逆合成分析方法出现之前曾在有机合成领域发挥重要作用,既包括各类化学结构的有机合成通法,又包括官能团的形成、转换或保护等合成反应。对于有明显化学结构特征和官能团的化合物,可考虑采用这种方法进行合成路线设计。
抗真菌药物克霉唑(clotrimazole,2-49)的关键中间体邻氯苯基二苯基氯甲烷(2-50)具有多种合成方法:首先可参考叔醇的合成方法,采用邻氯苯甲酸乙酯(2-51)与溴苯进行Grignard反应,先合成叔醇(2-52),再氯化得到(图2-22)。
图2-22 应用Grignard反应合成克霉唑关键中间体邻氯苯基二苯基氯甲烷
此种合成方法得到的邻氯苯基二苯基氯甲烷(2-50)质量较好,但由于应用了Grignard试剂,因此需要严格的无水条件,对原辅料质量要求比较严格;同时由于使用的溶剂乙醚易燃、易爆,故要求使用的反应设备要有严格的安全措施,使大规模生产受到一定限制。
鉴于此,参考四氯化碳与苯通过Friedel-Crafts反应可生成三苯基氯甲烷(2-53)的类型反应,人们又设计了以邻氯苯基三氯甲烷(2-56)为关键中间体的合成路线。
这种方法合成路线简短,原辅料来源方便,曾为工业化生产所采用。但在邻氯甲苯(2-54)经氯化反应制得邻氯苯基三氯甲烷(2-55)的过程中,一步反应需要引入三个氯原子,反应温度较高,反应时间较长,未反应的氯气易逸出,不易吸收完全,存在环境污染和设备腐蚀等严重问题(图2-23)。
第三种合成邻氯苯基二苯基氯甲烷(2-50)的方法是以邻氯苯甲酸(2-56)为原料,经两步氯化、两步Friedel-Crafts反应完成。这种方法虽然工艺路线较长,但原辅料来源方便,反应条件温和,各步反应产率较高,成本也较低,而且没有上述氯化反应的缺点,因此为工业化生产所广泛采用(图2-24)。
图2-23 应用Friedel-Crafts反应合成克霉唑关键中间体邻氯苯基二苯基氯甲烷
图2-24 联合应用氯化与Friedel-Crafts反应合成克霉唑关键中间体邻氯苯基二苯基氯甲烷
应用类型反应法进行药物或中间体的工艺路线的设计过程中,如果官能团的形成与转化等单元反应的排列方式可能出现两种或两种以上的不同方式时,不仅要从理论上考虑排列顺序的合理性,而且要从实际情况出发,着眼于原辅料、设备条件等因素,通过实验反复比较确定。化学反应类型相同,但进行顺序不同,则所应用的原辅料不同;原辅料不同,即反应物料的化学组成与理化性质不同,将导致反应的难易程度和反应条件不同,因此往往带来完全不同的结果:药物质量、收率、“三废”治理、反应设备和生产周期等方面都会有较大差异。
例如,β-受体阻断剂塞利洛尔(celiprolol,2-57)的合成(图2-25),对氨基苯乙醚(2-58)与 N , N -二乙胺基甲酰氯(2-59)作用,生成 N -酰化物(2-60),降低了的氨基的邻、对位定位作用,同时增大了氨基邻位空间位阻,使后面进行的Friedel-Crafts酰化反应发生在酚羟基的邻位,得到2-61;若先进行Friedel-Crafts反应, N -酰化反应居后,则乙酰化反应可同时发生在酚羟基和氨基的邻位,产物复杂。
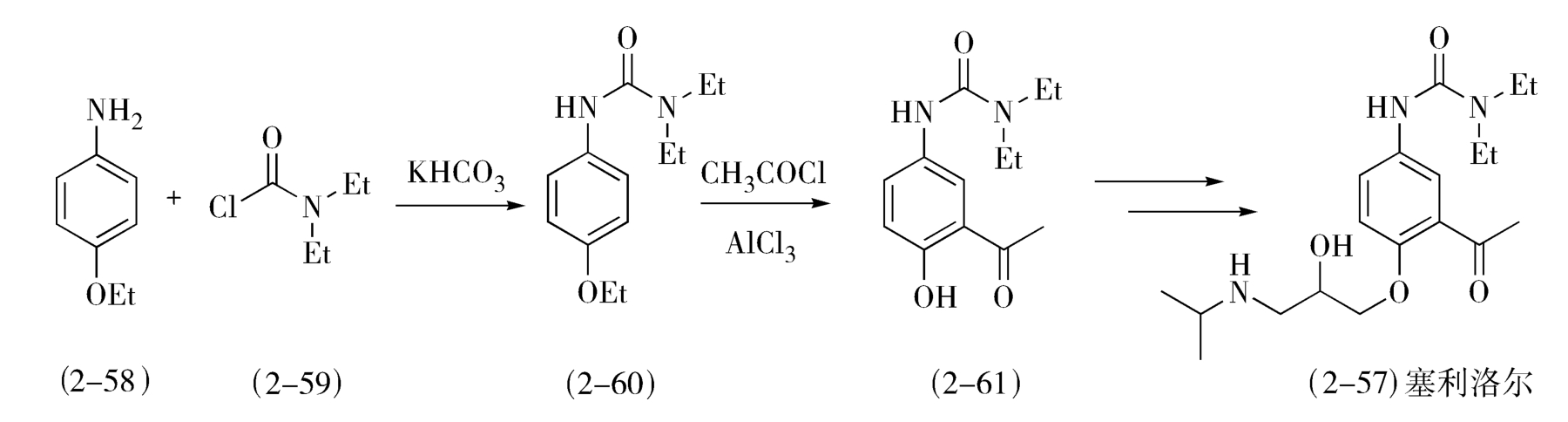
图2-25 塞利洛尔的合成
对于化学结构复杂、合成路线设计困难的药物,可模拟类似化合物的合成方法进行合成设计——实际上从初步的合成设想开始,通过文献调研,改进他人尚不完善的概念和方法来进行药物工艺路线设计,是药物合成设计中最为广泛使用和实用的方法。
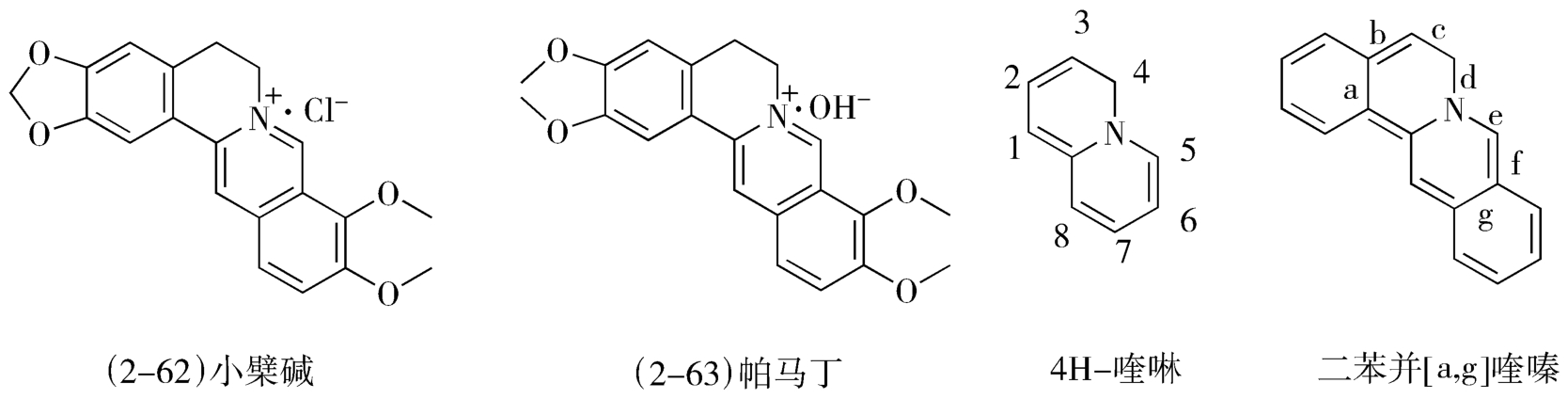
1.小檗碱的合成 中药黄连中的抗菌有效成分小檗碱(黄连素,berberine,2-62)与镇痛药帕马丁(palmatine,2-63)都是具有母核二苯并[a,g]喹嗪、含有稠合的异喹啉环结构的化合物。
(1)小檗碱的全合成 小檗碱(2-62)以3,4-二甲氧基苯乙酸(2-64)为起始原料,采用合成异喹啉环的方法,经Bischler-Napieralski反应及Pictet-Spengler反应先后两次环合全合成(图2-26)。
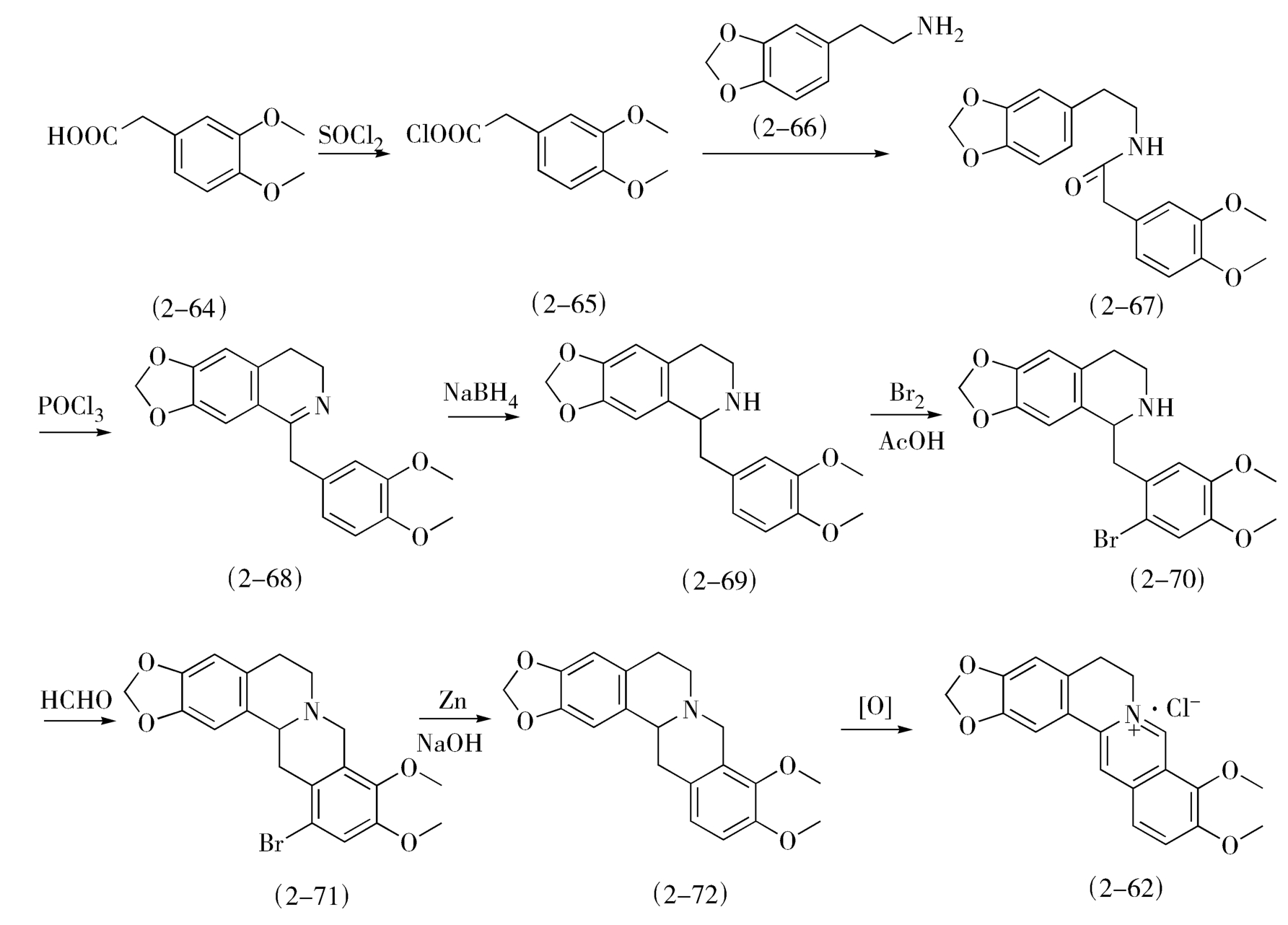
图2-26 小檗碱的全合成方法
在Pictet-Spengler环合反应前进行溴化,是为了提高环合位置的选择性,最后一步氧化反应可采用电解氧化或HgI做氧化剂。从合成化学的观点考察,这条路线合理可行,但由于路线较长,收率不高,且使用试剂昂贵,因而不适于工业化生产。
(2)模拟帕马丁的合成方法 1969年Muller等人发表了帕马丁(2-63)的合成方法:使用3,4-二甲氧基苯乙胺(2-73)与2,3-二甲氧基苯甲醛(2-74)进行脱水缩合生成Schiff碱(2-75),并立即将其双键还原转变成苯乙基苯甲基亚胺的骨架(2-76);然后与乙二醛反应,一次引进两个碳原子而合成二苯并[a,g]喹嗪环,按此合成途径得到二氢帕马丁高氯酸盐(2-77)与帕马丁高氯酸盐(2-63)混合物(图2-27)。
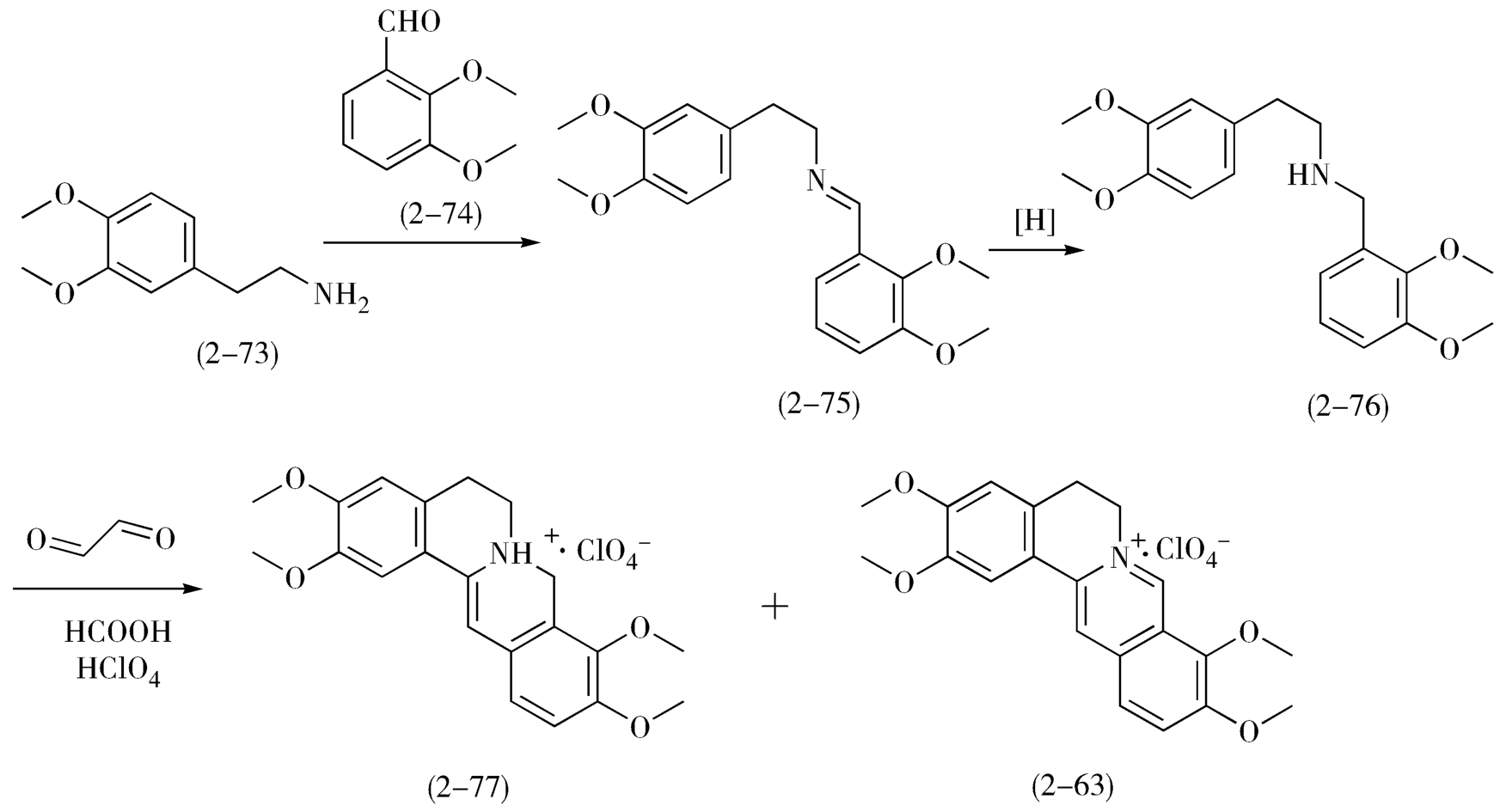
图2-27 帕马丁的合成方法
参照上述帕马丁的合成方法,从胡椒乙胺(2-66)与2,3-二甲氧基苯甲醛(2-74)出发,可快速合成小檗碱(2-62)(图2-28)。
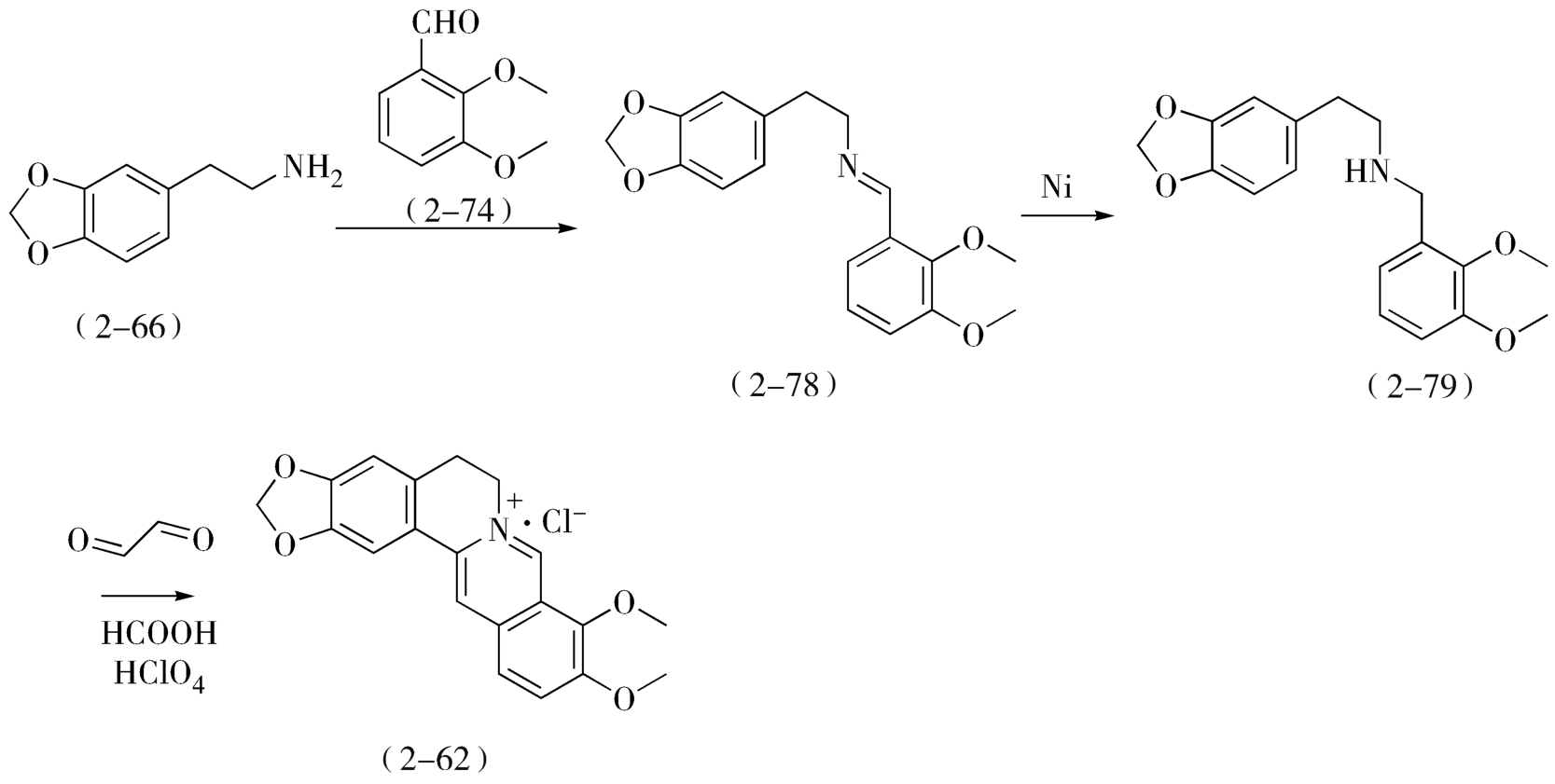
图2-28 模仿帕马丁的小檗碱合成方法
按此工艺路线制得的小檗碱(2-62)不含二氢化衍生物,产物的理化性质与抑菌能力同天然提取的黄连素完全一致,符合《中国药典》要求,并较前述路线更为简捷,所用原料2,3-二甲氧基苯甲醛(2-74)是工业生产香兰素的副产物,因此较适于大规模生产。
应用模拟类推法的要点在于适当的类比和对有关化学反应的了解,使用时应注意比较已有合成方法、类似化学结构及化学活性的差异。
2.喹诺酮类抗菌药物的合成方法 喹诺酮类抗菌药物(2-80~2-86)具有相似的基本骨架,合成多以多取代苯胺为起始原料构建吡酮酸环。构建方法是在诺氟沙星(norfloxacin,2-80)和环氧沙星(ciprofloxacin,2-81)等早期品种的合成基础上发展而来的,主要有取代苯胺与乙氧基亚甲基丙二酸二乙酯(EMME,2-87)缩合成环和经取代芳胺环上的亲核取代反应成环两种。

(1)取代苯胺与EMME缩合成环(图2-29)。
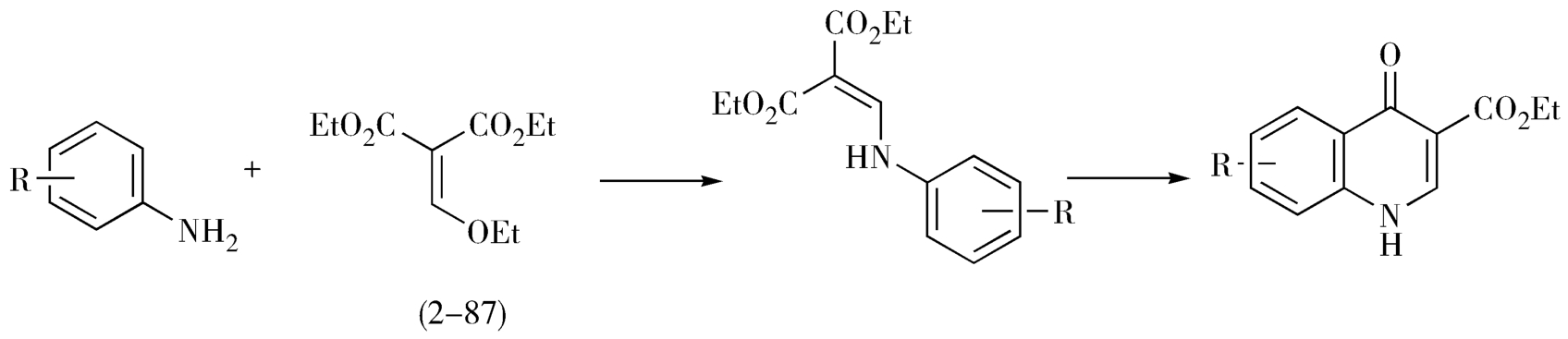
图2-29 取代苯胺与EMME缩合成吡酮酸环
诺氟沙星的合成以3-氯-4-氟苯胺(2-88)为原料,先与EMME脱乙醇缩合,然后在250~260℃加热环合形成吡酮酸结构的2-89,溴乙烷为烃化试剂完成N原子上的乙基化,然后水解,引入哌嗪基,得到诺氟沙星(2-80)(图2-30)。
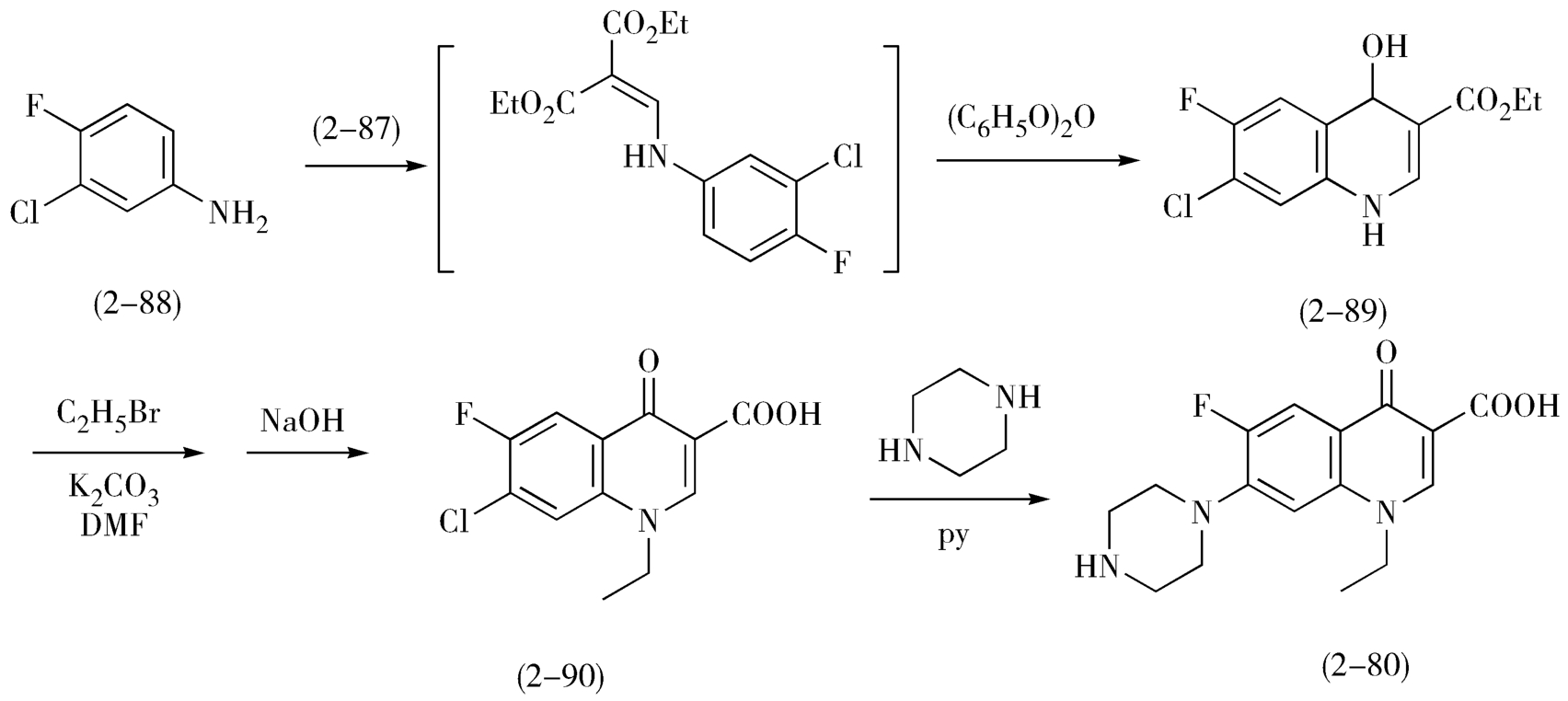
图2-30 诺氟沙星的合成
与诺氟沙星的合成类似,氟罗沙星(fleroxacin,2-84)可以2,3,4-三氟硝基苯(2-91)为起始原料,经还原、与EMME缩合、高温环合、氟乙基化、引入 N -甲基哌嗪、酸水解等6步反应得到(图2-31)。
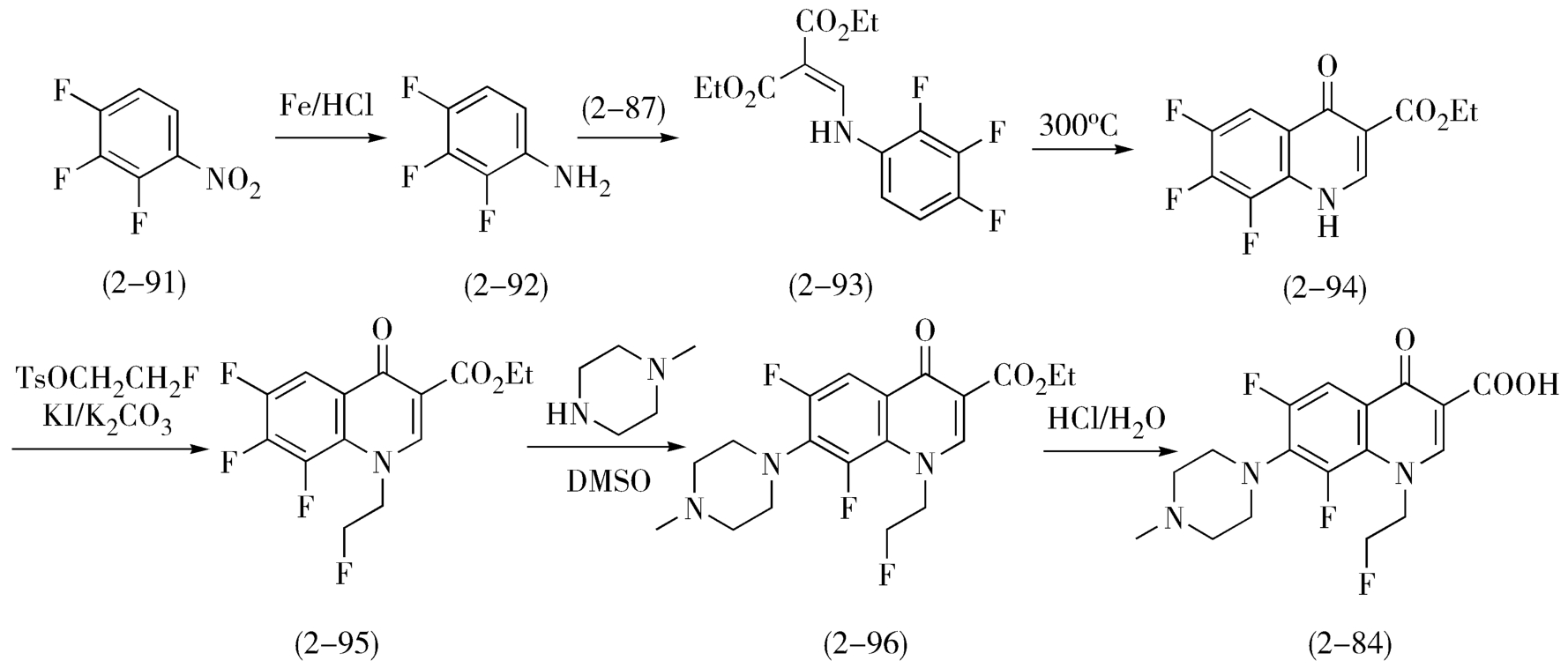
图2-31 氟罗沙星的合成
(2)经取代芳胺环上的亲核取代反应成环,离去基团为卤素或硝基(图2-32)。
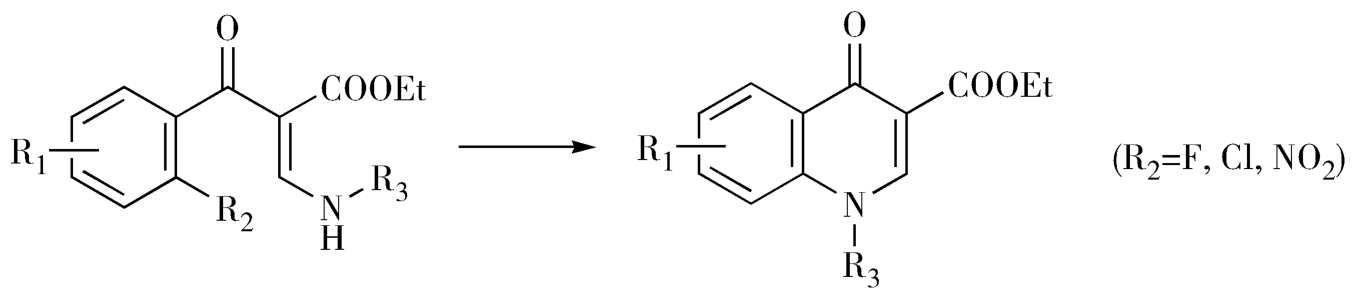
图2-32 经亲核取代反应合成吡酮酸环
环丙沙星(2-81)与诺氟沙星(2-80)的结构差异在于1位取代基分别为环丙基和乙基,但两者的合成路线却有很大不同。环丙沙星的合成从2,4-二氯-5-氟苯甲酸(2-97)开始,成酰氯后与β-环丙胺丙烯酸甲酯(2-99)缩合,环合、水解,再引入哌嗪基(图2-33)。
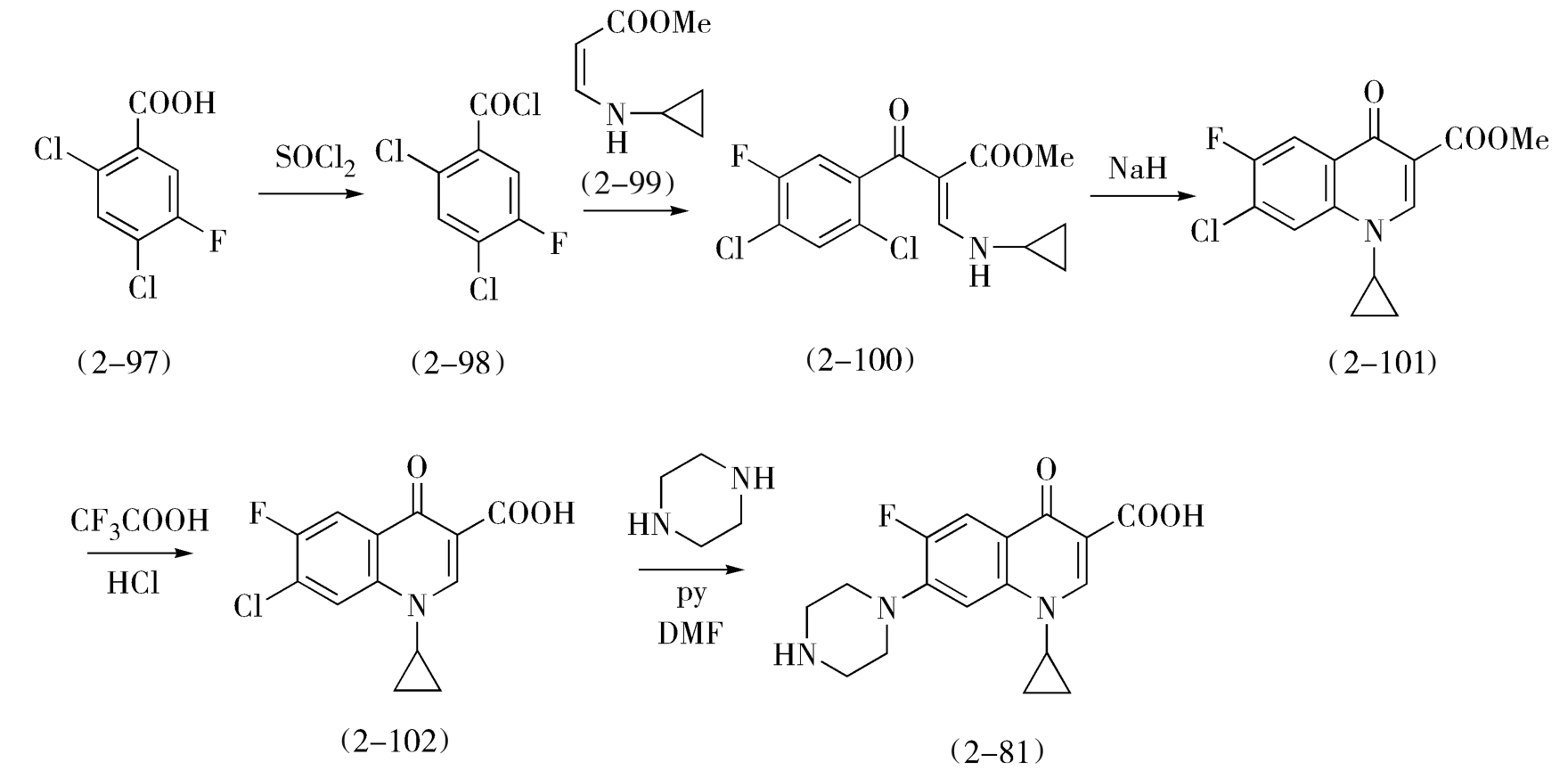
图2-33 环丙沙星的合成
参照上述环丙沙星的合成方法,加替沙星(2-85)可以2,4,5-三氟-3-甲氧基苯甲酸(2-103)为起始原料,成酰氯后与3-二甲氨基丙烯酸乙酯(2-105)反应后,再用环丙胺(2-106)置换,取代关环后得到关键中间体(2-108)。这时若将2-108水解直接与2-甲基哌嗪缩合,由于8位甲氧基的强推电子作用,使得7位氟作为亲核取代反应的离去基团活性大大降低,故缩合收率仅为19.4%;因此将2-108先与硼化物反应生成络合物(2-109),由于4位羰基上氧原子的p电子向硼原子的空轨道发生转移,使得4位羰基的吸电子效应进一步增强,从而提高了7位氟对亲核试剂的反应活性,与2-甲基哌嗪缩合,然后水解得到加替沙星,按此路线改进后缩合与水解两步产率可提高到75.5%(图2-34)。
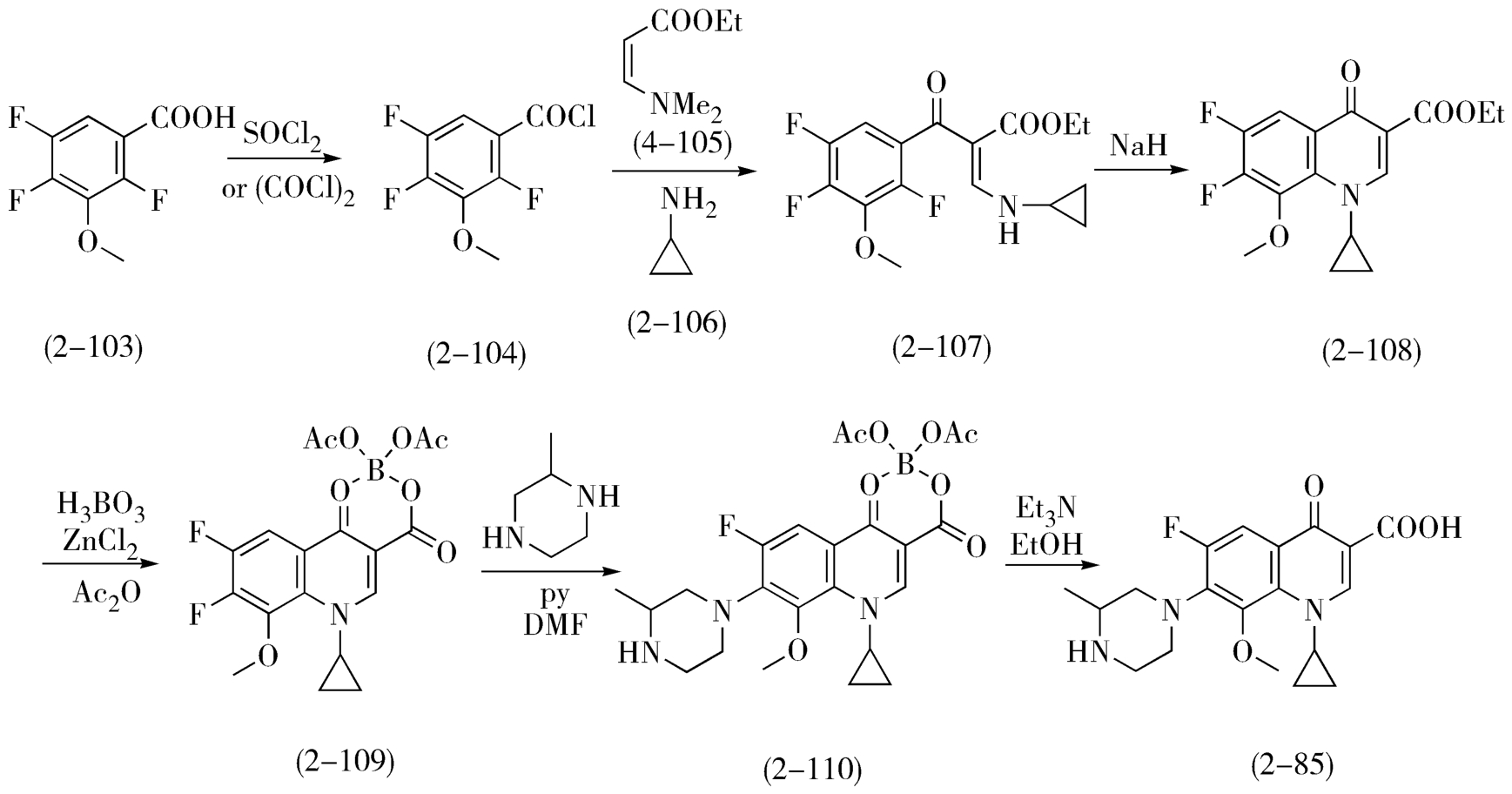
图2-34 加替沙星的合成
(3)喹诺酮类抗菌药物合成方法的改进:上述合成喹诺酮类抗菌药物的两条工艺路线都比较成熟,但工艺步骤相对较多,特别是都将7位引入哌嗪基的反应放在路线的最后,且收率较低,技术难度较大。近年来人们对喹诺酮类抗菌药物的合成又有新的改进,尤其是对成环工艺改进很大:以2-氯-4-氨基-5-氟苯甲酸乙酯(2-111)为起始原料,后4步总收率可达94.5%(图2-35)。
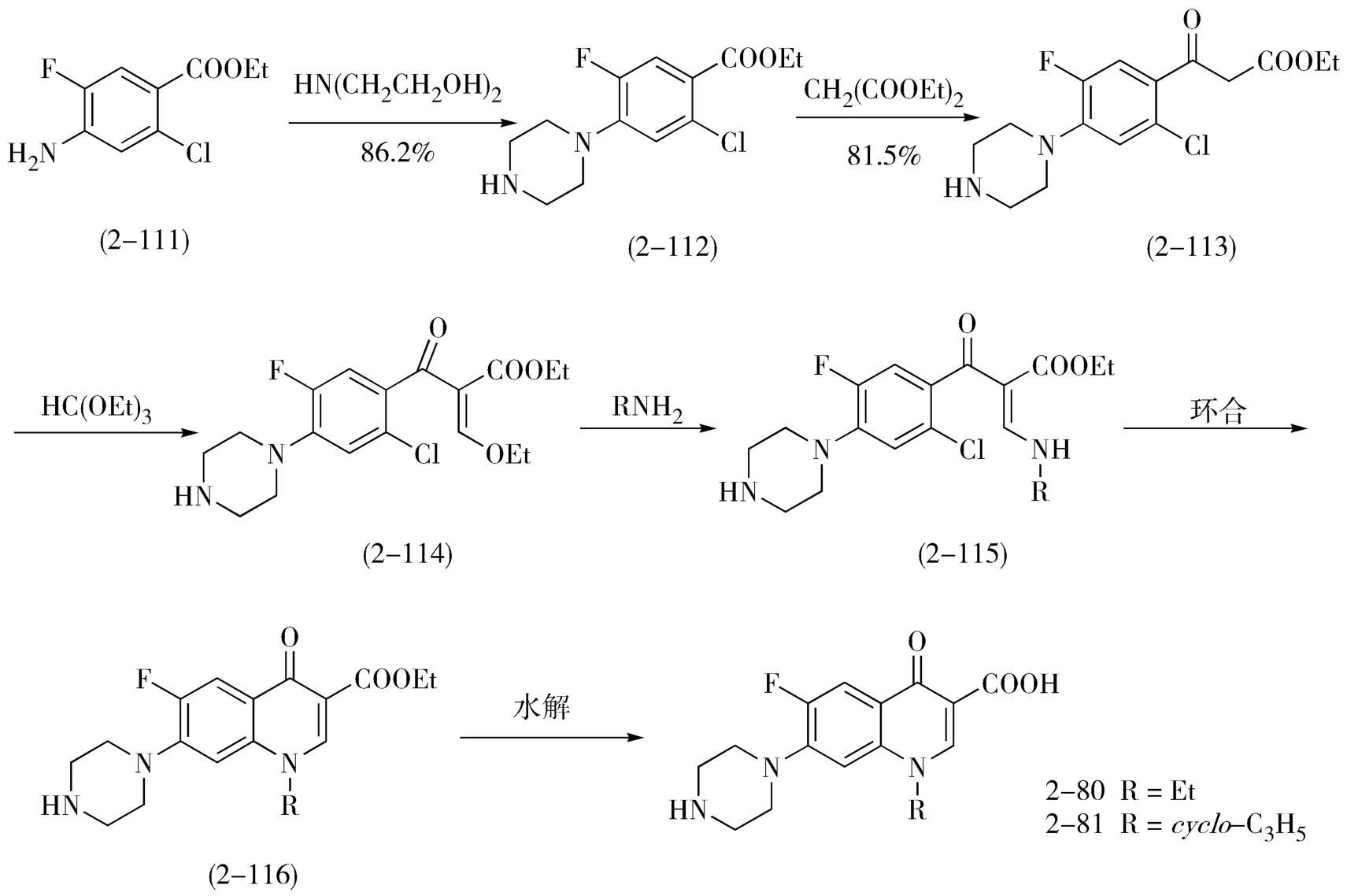
图2-35 喹诺酮类抗菌药物的工艺改进
3.质子泵抑制剂的合成方法 质子泵抑制剂(PPI)为H + /K + -ATP酶抑制剂,是最新一类抑制胃酸分泌的药物。这是一类以吡啶甲基亚硫酰基苯并咪唑为基础结构的化合物,目前的质子泵抑制剂是在奥美拉唑(omeprazole,2-117)的合成基础上发展而来的,这些质子泵抑制剂通常采用氧化相应的硫醚化合物来制备,得到的是外消旋混合物,采用特殊氧化方法(如加入手性试剂),可获得单一对映体或富含对映体形式的产物。

奥美拉唑的合成以2,3,5-三甲基吡啶(2-122)为原料,通过氧化、硝化、甲氧基化、在乙酐中重排、水解、氯化亚砜氯化,与2-巯基-5-甲氧基苯并咪唑在甲醇钠参与下缩合成硫醚,最后再经间氯过氧苯甲酸(m-CPBA)氧化制得(图2-36)。
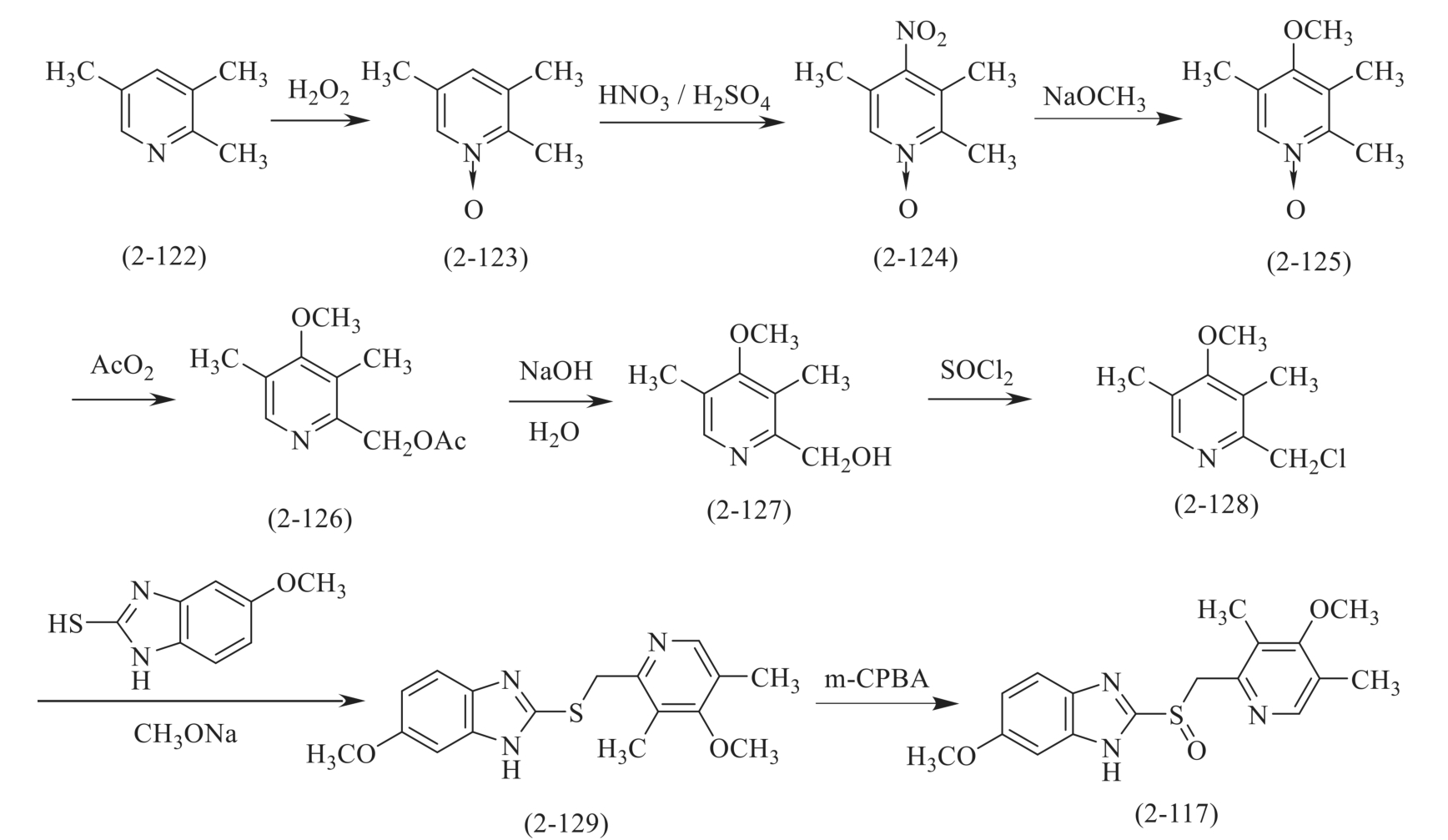
图2-36 奥美拉唑的合成
与奥美拉唑的合成类似,兰索拉唑(lansoprazole,2-118)可以2,3-二甲基吡啶(2-130)为起始原料,经氧化、硝化、在乙酐中重排、水解、氯代、取代,最后m-CPBA氧化等7步反应等到(图2-37)。
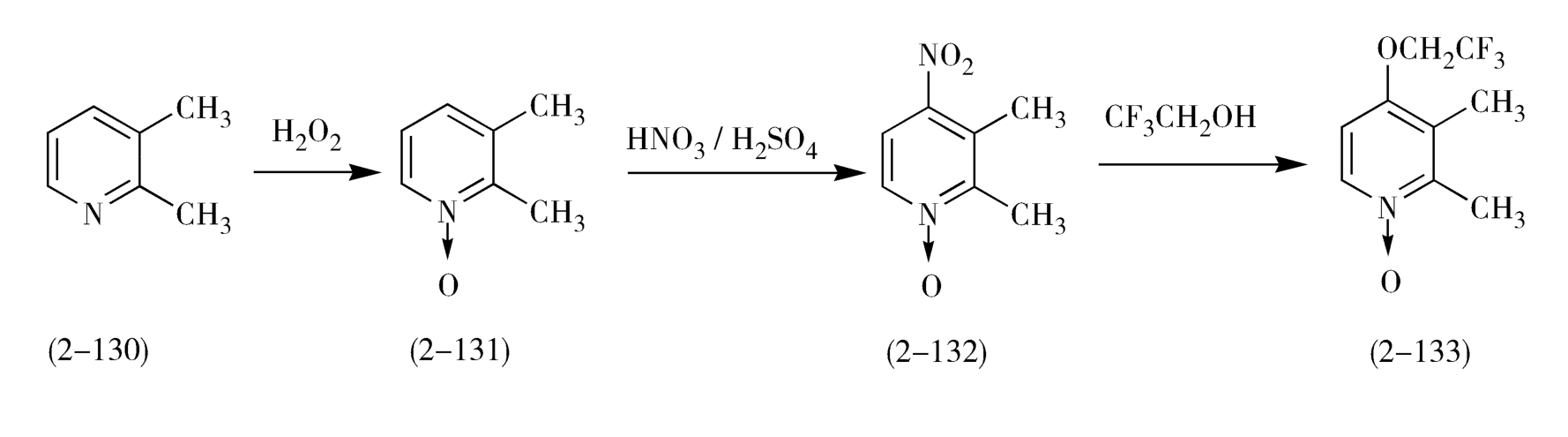
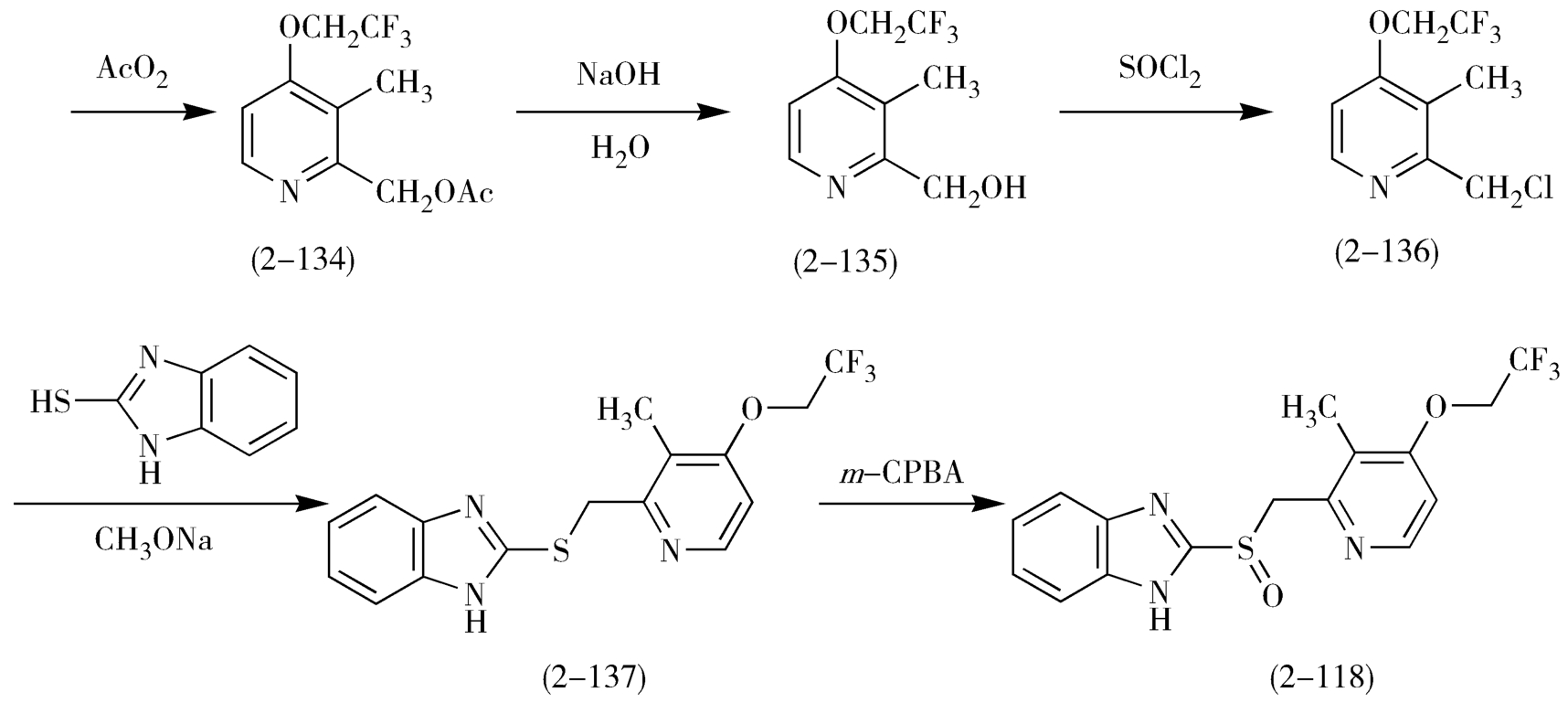
图2-37 兰索拉唑的合成
泮托拉唑(pantoprazole,2-119)的合成同样也是参照奥美拉唑的合成方法,以2-甲基-3-甲氧基-4-氯吡啶(2-138)为原料,经双氧水氧化得到N-氧化物(2-139),然后经甲氧基化反应、重排反应、水解反应、氯化反应及缩合反应得到泮托拉唑(图2-38)。
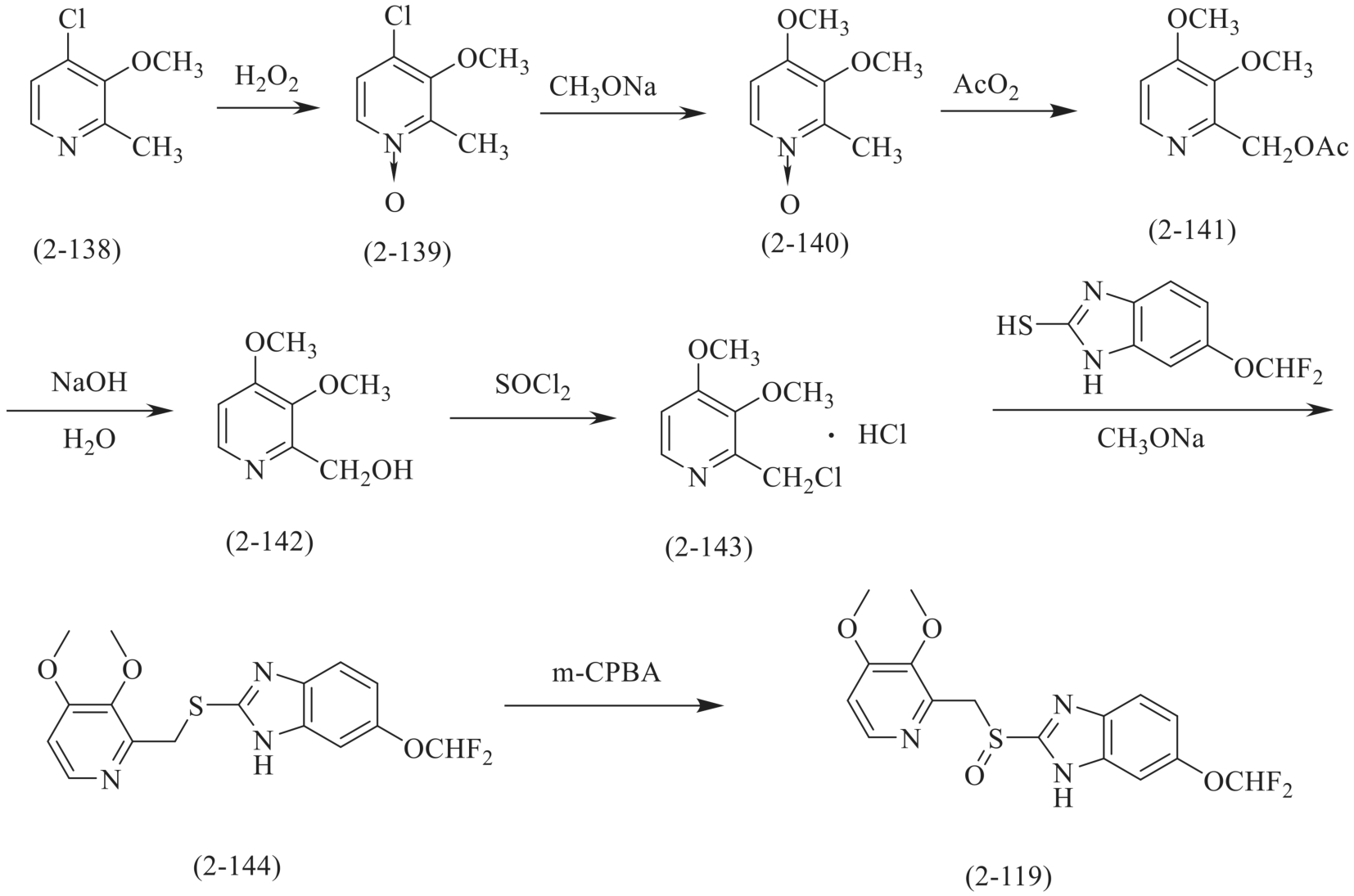
图2-38 泮托拉唑的合成
雷贝拉唑(rabeprazole,2-120)的合成原料与兰索拉唑相同,以2,3-二甲基吡啶(2-130)为原料,经过氧化、硝化、重排、水解、取代等反应,经过五个化合物的合成过渡,再与2-巯基-1 H -苯并咪唑缩合,制得关键中间体2-[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基硫-1 H -苯并咪唑(2-148),最终选择合适的氧化剂制得雷贝拉唑(图2-39)。
由于取代基的不同,各个药物的药理作用差别较大,在结构中有硫原子连接的不对称亚砜结构,存在一个手性中心,因此使得每个化合物都有一对对映体,各对映体的药理活性在人体内表现不同,单一异构体具有作用范围广、药效好、血浆半衰期长、副作用小等特点。
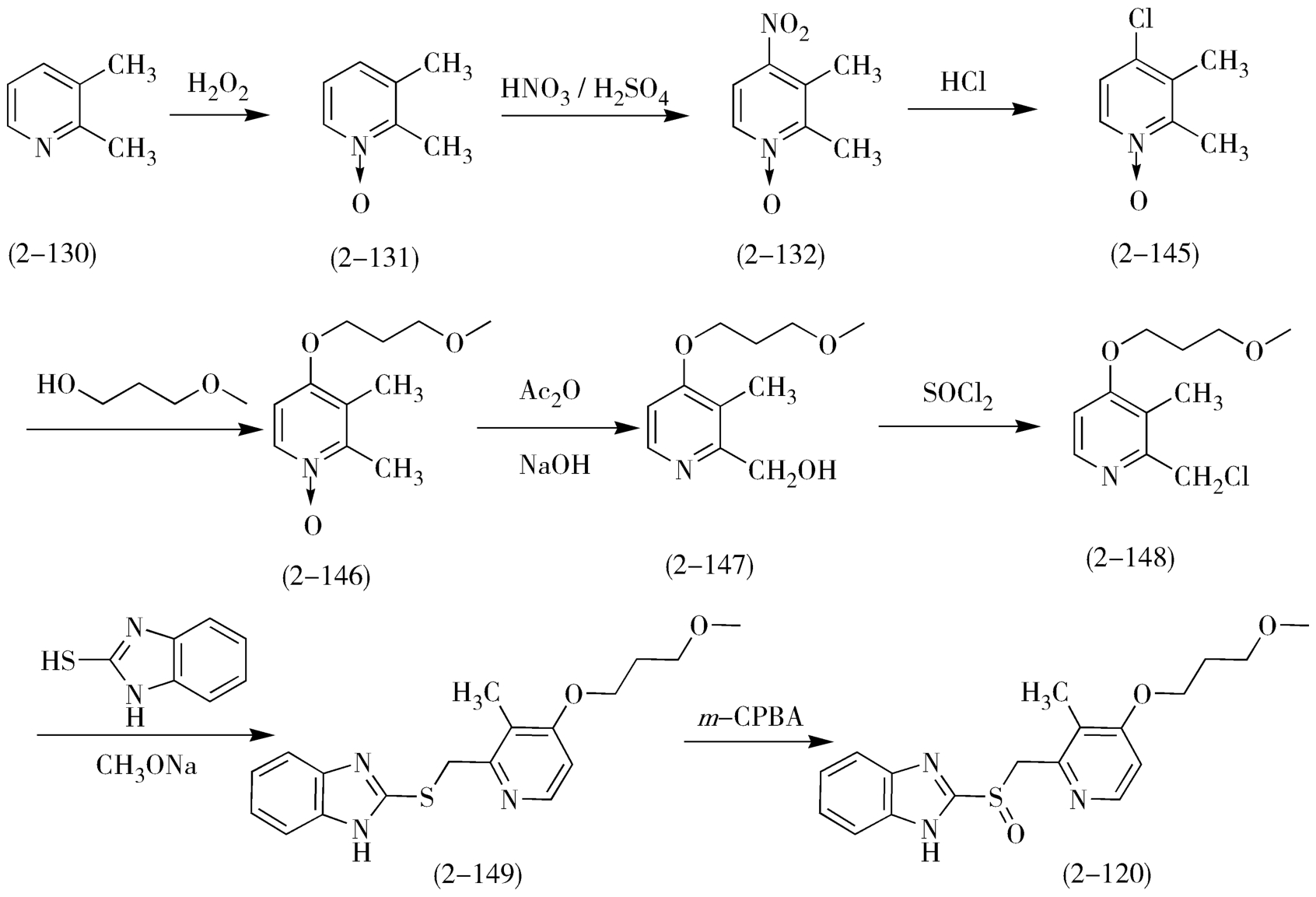
图2-39 雷贝拉唑的合成
埃索美拉唑(esomeprazole,2-121)是第一个上市的光学纯质子泵抑制剂,为奥美拉唑的左旋异构体,对消化性溃疡的治疗效果强于消旋体3~5倍,主要有3种合成路线:手性拆分法、生物氧化法和不对称氧化法。手性拆分法是以消旋奥美拉唑为原料,借助一种手性试剂或者特异微生物,实现消旋体的分离。生物氧化法是用生物酶对奥美拉唑硫醚进行氧化或对奥美拉唑砜进行还原制得埃索美拉唑。不对称氧化法是直接用手性催化剂进行催化氧化奥美拉唑硫醚制得的奥美拉唑的左旋单一体。
手性拆分法很难有效彻底的对消旋体奥美拉唑进行拆分,并且该方法会损失一半的原料,成本较高,从而使工业化生产受限制(图2-40)。
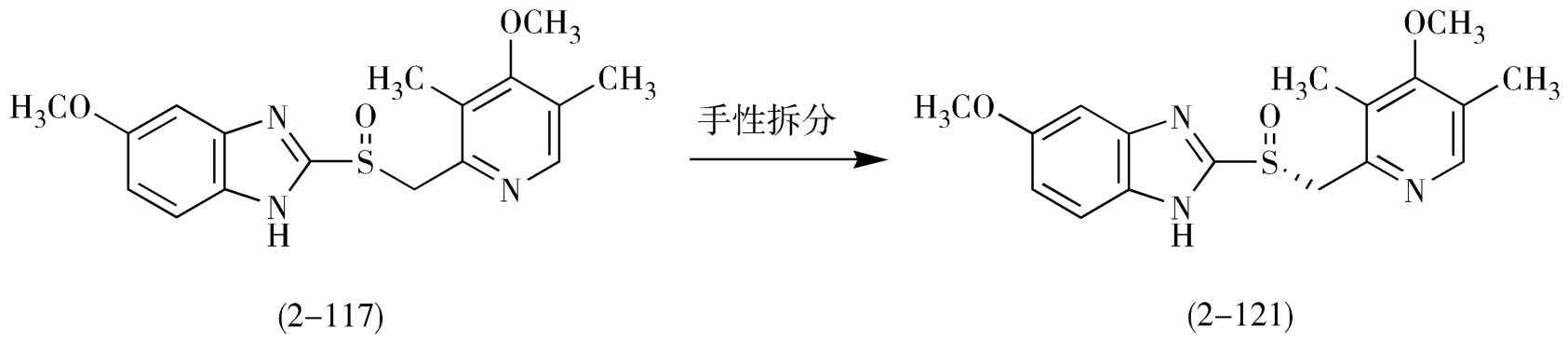
图2-40 手性拆分反应路线图
生物氧化法氧化选择性较低、副产物较多、工艺稳定性差,对反应条件和设备要求较高,操作繁琐,也不适用于工业化生产。
目前采用的合成工艺主要是不对称氧化法,先制备中间体前手性硫醚物(2-129),然后在手性环境中用氧化剂对中间体(2-129)进行不对称氧化,产品收率高,手性纯度好,避免了采用拆分法中拆分试剂价格昂贵、物料损失大等缺点,氧化反应条件温和,方便可控,适合工业化生产(图2-41)。
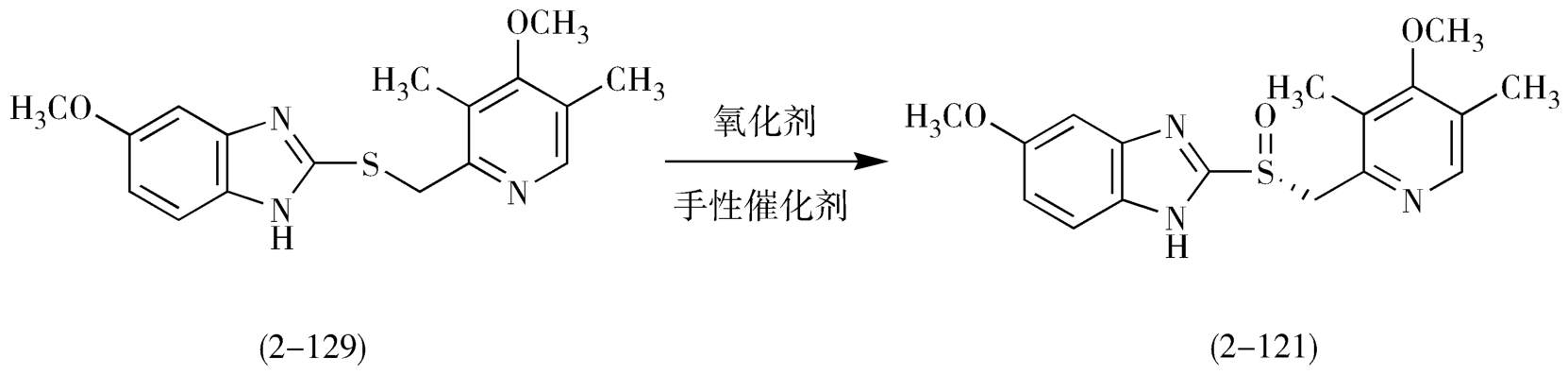
图2-41 不对称催化氧化反应路线图
随着开发单一手性质子泵抑制剂逐渐成为热门,除了埃索美拉唑之外,左旋泮托拉唑、右旋雷贝拉唑等也是采用不对称氧化法合成的,这些单一异构体的合成及应用也将会为消化系统药物开创另一个局面(图2-42、图2-43)。
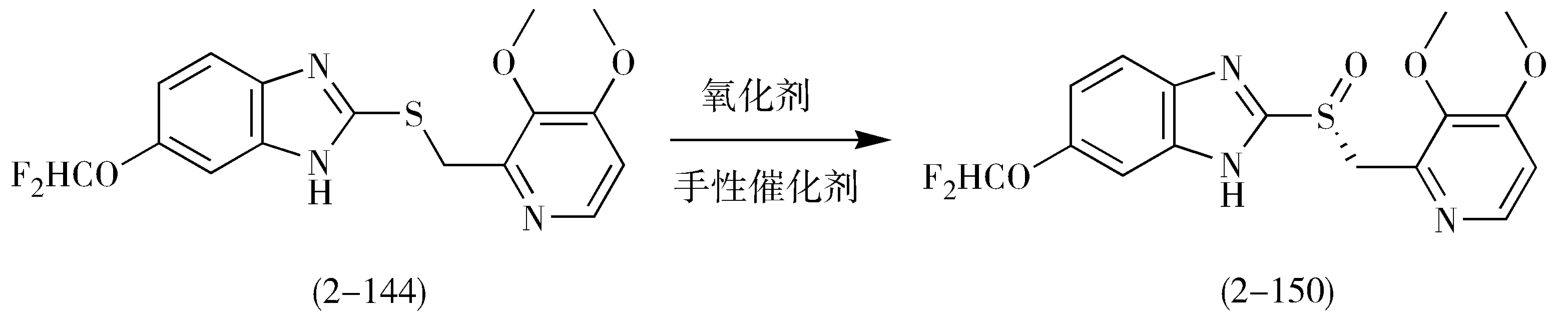
图2-42 左旋泮托拉唑不对称催化氧化反应路线图
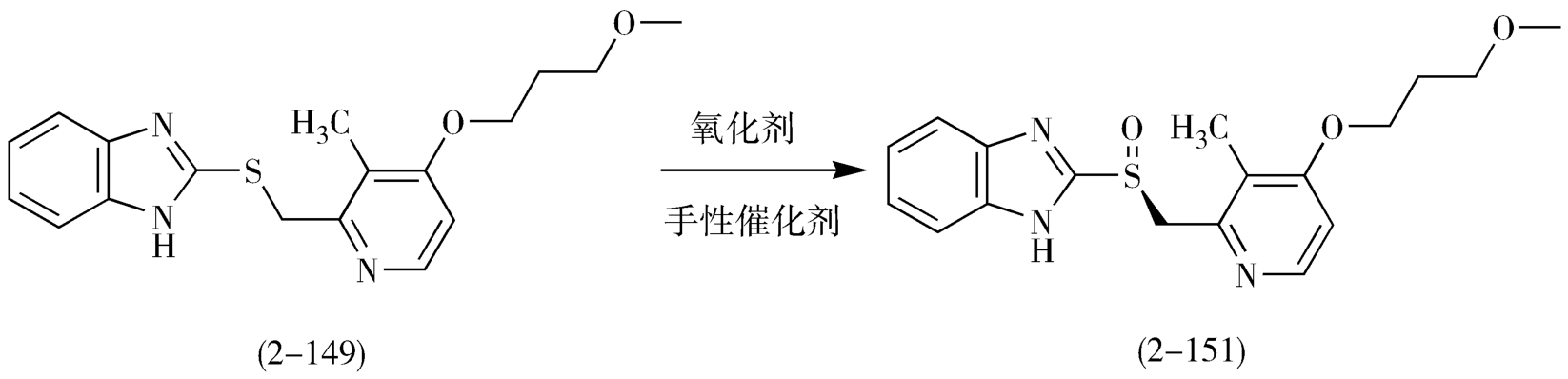
图2-43 右旋雷贝拉唑不对称催化氧化反应路线图