
下载掌阅APP,畅读海量书库
立即打开



逆合成法是药物生产工艺路线设计的最基本的方法,也叫作反合成法(antithetic synthesis),其他一些更为复杂的设计方法都是建立在此方法基础上的,所以要先掌握逆合成法。逆合成法的整个设计思路也被称为逆(反)合成分析,即从目标分子的结构出发,逐步考虑,层层分解,先考虑由哪些中间体合成目标物,再考虑由哪些原料合成中间体,最后的原料就是起始物(起始原料,startingmaterial,SM)。
进行一个药物分子的合成设计工作往往需要三个步骤,即分子考察、逆合成分析和正向检查。这三个步骤各有侧重,后者以前者为依据,具有严密的逻辑关系。
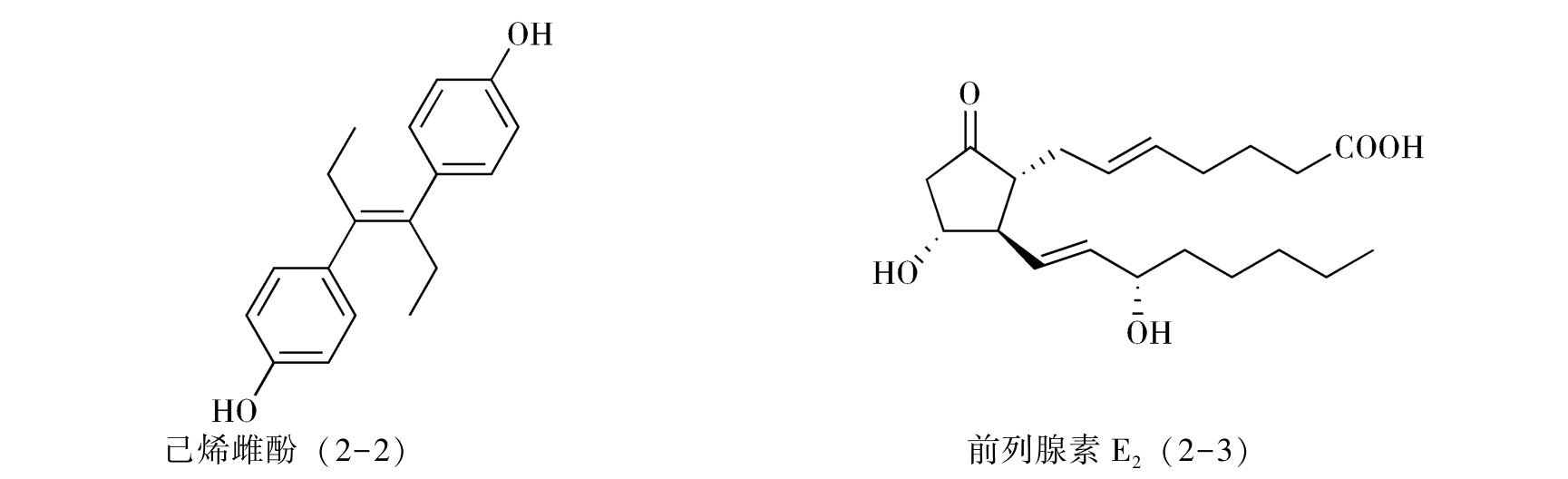
1.分子考察 考虑对一个特定药物进行合成,第一步是对这个药物分子的结构特征和理化性质进行收集和考察,由此可以简化合成中的问题或避免不必要的弯路。例如,非甾体雌激素药物己烯雌酚(diethylstilbestrol,2-2)的分子带有明显的对称性,因此可以考虑只合成一部分结构单元,采用分子对接的方法合成目标药物分子,从而简化合成步骤(详见分子对称法);而在考虑前列腺素E 2 (2-3)的合成时,由于已知分子中β-羰基酮体系是不稳定的,因此可以安排在合成的最后几步形成这一结构单元,使其避免经历较多的化学反应。
分子考察是极为重要的阶段,通过分子考察可确定分子特有的骨架结构,从而得到基本的转换方式(见第四节“基于转换方式的合成策略”叙述)。
2.逆合成分析 进行药物分子合成的第二步是以上述分析为基础,从药物本身出发,一步步倒推出合成此药物的各种合成路线和起始原料,也就是我们通常所说的逆合成法(retrosynthesis)。逆合成分析过程要求:
(1)每步都有合理且合适的反应机理和合成方法。
(2)整个合成要做到最大可能的简单化。
(3)有被认可的(即市场能供应的)原料。
重复和交替使用转化过程,就可以推导出合成目标药物分子所需的起始原料。具体做法就是一步一步地进行逆合成分析,最终推导出合成此目标化合物的可能路线和易得的起始原料。每一步逆合成可以得出若干个合成子,由合成子再推导得到试剂或反应底物,如果此试剂或反应底物仍然难得,则再进行进一步的逆合成。
例如,局麻药物普鲁卡因(procaine,2-4)的转化分别经历了两次官能团的转化和一次分子骨架的转化,最终得到起始原料对硝基甲苯(2-8)(图2-3)。
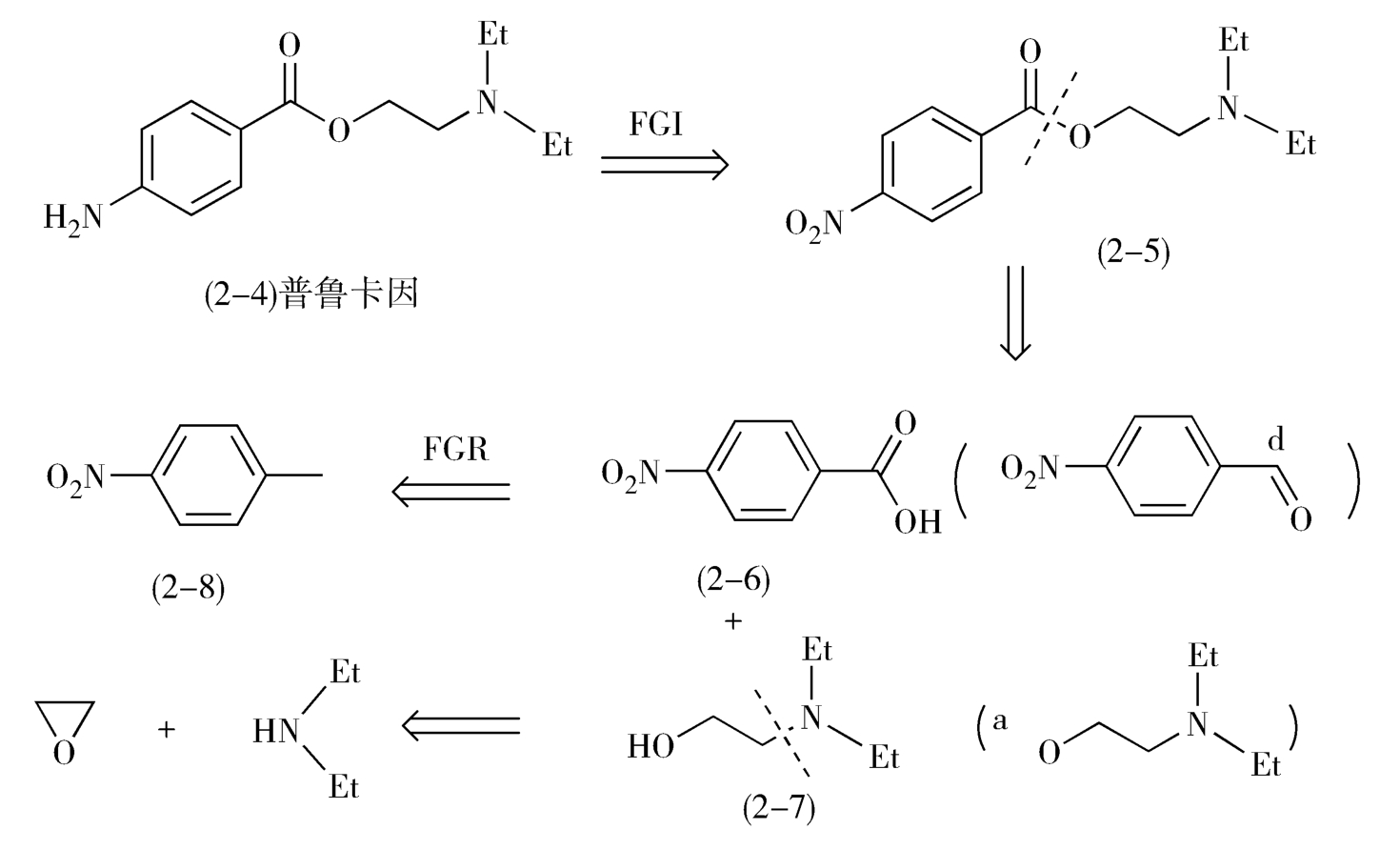
图2-3 普鲁卡因的逆合成分析
在合成普鲁卡因(2-4)的过程中,以重铬酸钠氧化对硝基甲苯(2-8),生成对硝基苯甲酸(2-6),再与二乙胺基乙醇(2-7)进行酯化反应,经二甲苯共沸脱水得硝基卡因(2-5),(2-5)再于稀盐酸中用铁粉还原即得产物(图2-4)。
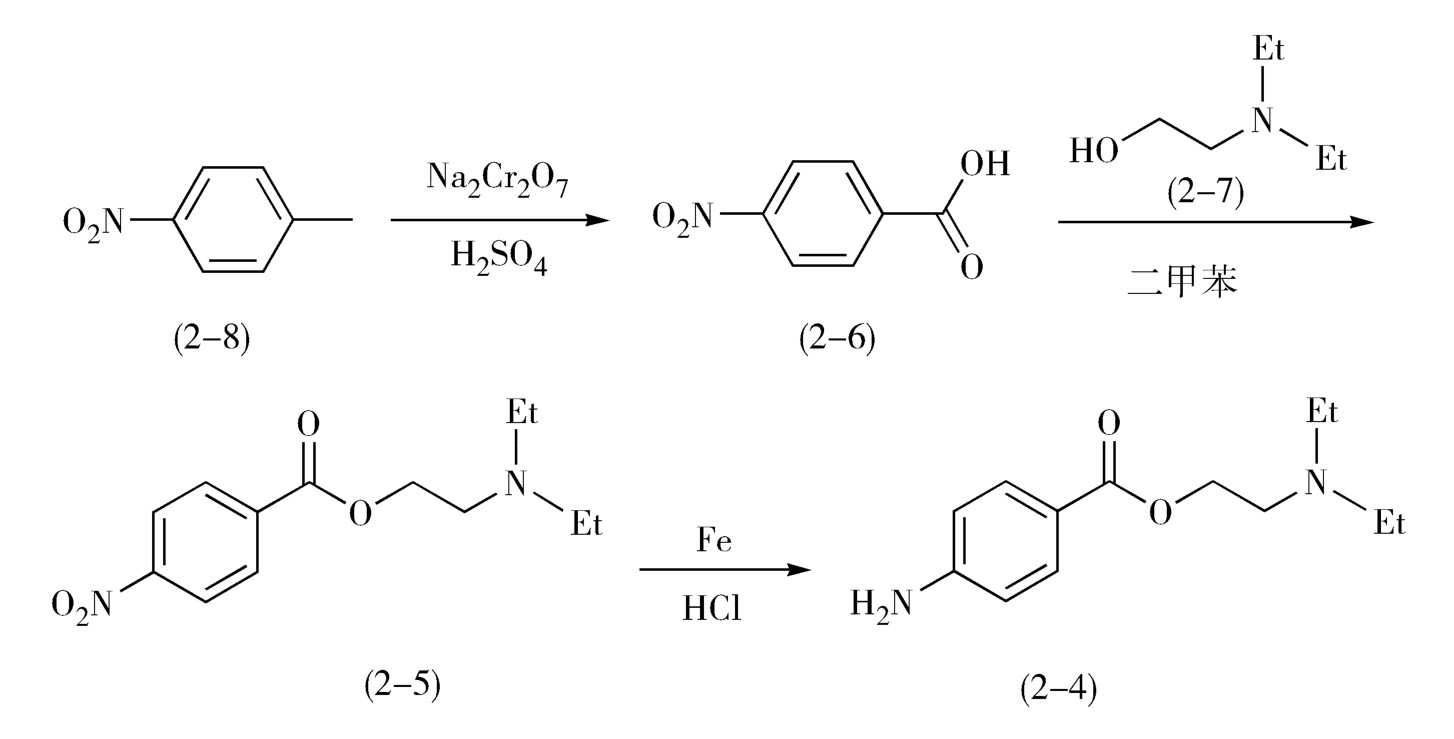
图2-4 普鲁卡因的合成
推导合成子的目的是为了合成设计服务,由于推导出的有些合成子所依据的转化、分拆还不存在相应的反应,因此一般没必要推导出所有可能的合成子。我们使用E.J.Corey本人使用过的例子来说明这个问题:
图2-5给出了目标分子的几种可能的分拆方式,由此得到一系列合成子:
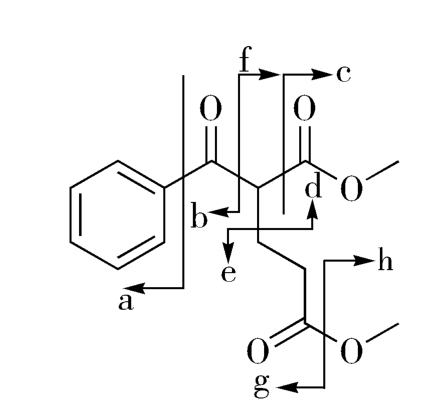
图2-5 目标分子(2-9)分拆的可能性
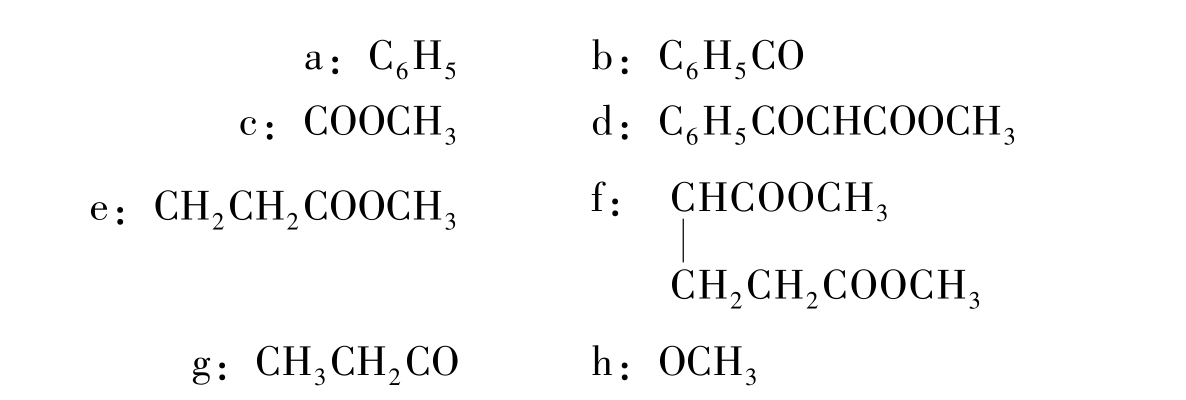
但这些推导出的合成子,仅有d和e具有实际应用价值,由两种合成子推导出的相应易得试剂经Michael加成,即得目标产物(图2-6)。
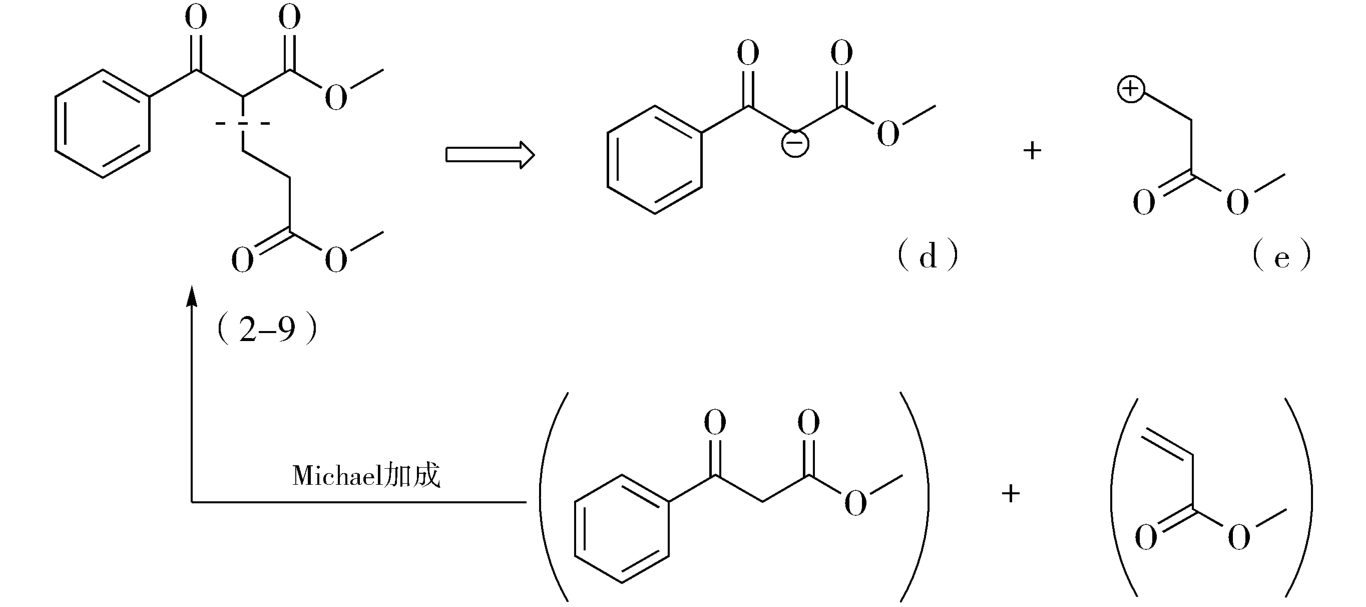
图2-6 目标分子(2-9)合成途径
从逆合成分析过程中得到实用的合成子和易得的中间体或原料,需要大量的实际工作经验,所幸的是有机合成化学家已从已知合成工作实践中总结了许多规律,可以作为我们药物合成设计的有益借鉴(见后“基于转换方式的合成策略”叙述)。
设计药物分子的合成路线是比较困难的问题,即使结构不太复杂的药物分子,在它们的合成过程中也总包含有骨架与官能团的变化,这样就产生了一个问题:在解决骨架与官能团都有变化的合成问题时应该先考虑什么。化合物的性质主要是由分子的官能团决定的,但是在解决骨架与官能团都有变化的合成问题时,要优先考虑骨架的形成,这是因为官能团是附着于骨架上的,骨架建立不起来,官能团就没有根基。
考虑骨架的形成时,首先研究目标分子的骨架是由哪些较小单元的骨架、通过哪些成键反应结合而成的,较小单元的骨架又是由哪些更小的碎片骨架、通过何种成键反应结合而成的。依此顺序推断下去,直到得出最小碎片的骨架,也就是应该使用原料的骨架。
但是考虑骨架形成的过程中又不能脱离官能团。碳骨架的形成和官能团的运用是两个不同的方面,两者相对独立但又相互联系:碳骨架只有通过官能团的运用才能装配——反应是在官能团上发生的,或是在由于官能团的影响而产生的活跃部位(如羰基或双键)上发生的,因此应时刻考虑官能团对整个分子结构的影响,特别是在建立碳-碳键之前,应先建立碳-杂键(碳-杂键基本是由于官能团反应形成的)。
3.正向检查 路线设计是药物合成工作的起始工作,也是最重要的一环。在对分子结构特征和理化性质收集、考察及进行逆合成分析,设计出初步的合成路线之后,路线设计的第三步工作就是在此逆合成分析设计的基础上进行正向检查,确定合成路线的切实可行性,这时主要是对合成所涉及的化学反应进行进一步的考查,保证合成路线的顺利完成。
逆合成分析过程如同数学运算,数学运算是从已知条件开始,最终获得正确答案,虽然解题的过程只要逻辑正确可以因人而异,却有繁简之分;而任何一条合成路线的设计,只要能合成出所需要的化合物,应该说都是合理的,但是合成的技巧、路线设计水平的高低却体现在路线的简洁、产率的高低、原料的来源方便与否、操作的难易等诸多方面。为了设计一条高水平的合成路线,应该科学、合理地做好逆合成分析工作。一般来说,逆合成分析工作应遵循以下顺序。
1.由目标分子结构和反应性决定逆合成顺序 在进行药物分子合成过程中,首先需要对目标分子有充分认识,并对其反应深入了解,通过对目标分子的结构考察,分析其结构特征及化学反应性质,从而设计出有针对性的合成路线。在目标分子的分拆过程中,应先分拆对称部分(见分子对称法);然后分拆分子中不稳定部分或影响分子反应性及选择性的部分。
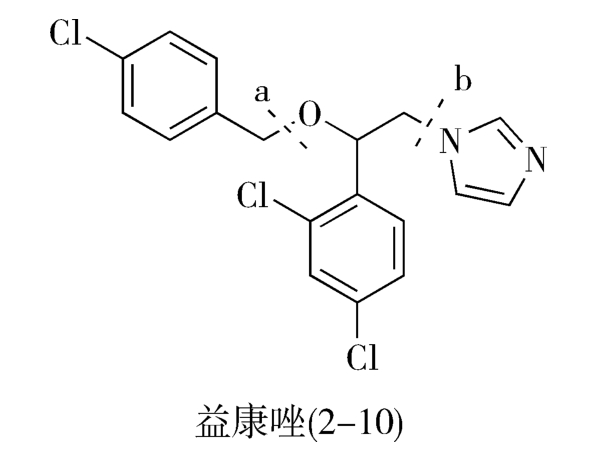
目标分子中C—N、C—S、C—O等碳-杂键通常是该分子的拆键部位,即分子的连接部位。例如,抗真菌药益康唑(econazole,2-10)分子中有C—O和C—N两个拆键部位,可从这两处追溯其合成的前一步中间体:
如图2-7所示,从虚线a处断开C—O键,益康唑的前体为对氯甲基氯苯(2-13)和1-(2,4-二氯苯基)-2-(1-咪唑基)乙醇(2-11);2-11继续分拆,得到1-(2,4-二氯苯基)-2-氯代乙醇(2-15)和咪唑(2-14)。
从虚线b处断开C—N,前体为咪唑(2-14)和2-(4-氯苯甲氧基)-2-(2,4-二氯苯基)氯乙烷(2-12)。2-12进一步分拆,前体为对氯甲基氯苯(2-13)和1-(2,4-二氯苯基)-2-氯代乙醇(2-15)。
综上所述,无论按照a途径或b途径分拆,得到的合成益康唑的基本原料都是对氯甲基氯苯(2-13)、咪唑(2-14)和1-(2,4-二氯苯基)-2-氯代乙醇(2-15),问题是先合成C—O键还是先合成C—N键有利呢(注意:合成与分拆的顺序相反!)
按照b途径分拆,1-(2,4-二氯苯基)-2-氯代乙醇(2-15)与对氯甲基氯苯(2-13)在碱性条件下制备中间体2-12时,理论基础是Williamson醚合成,但是由于2-15自身也存在活性的伯氯基团,所以不可避免地将发生2-15自身分子的烷基化反应,从而使反应复杂化,降低2-12的收率。因此,先形成C—N键,再形成C—O键的a途径对合成益康唑分子更为有利(图2-7)。
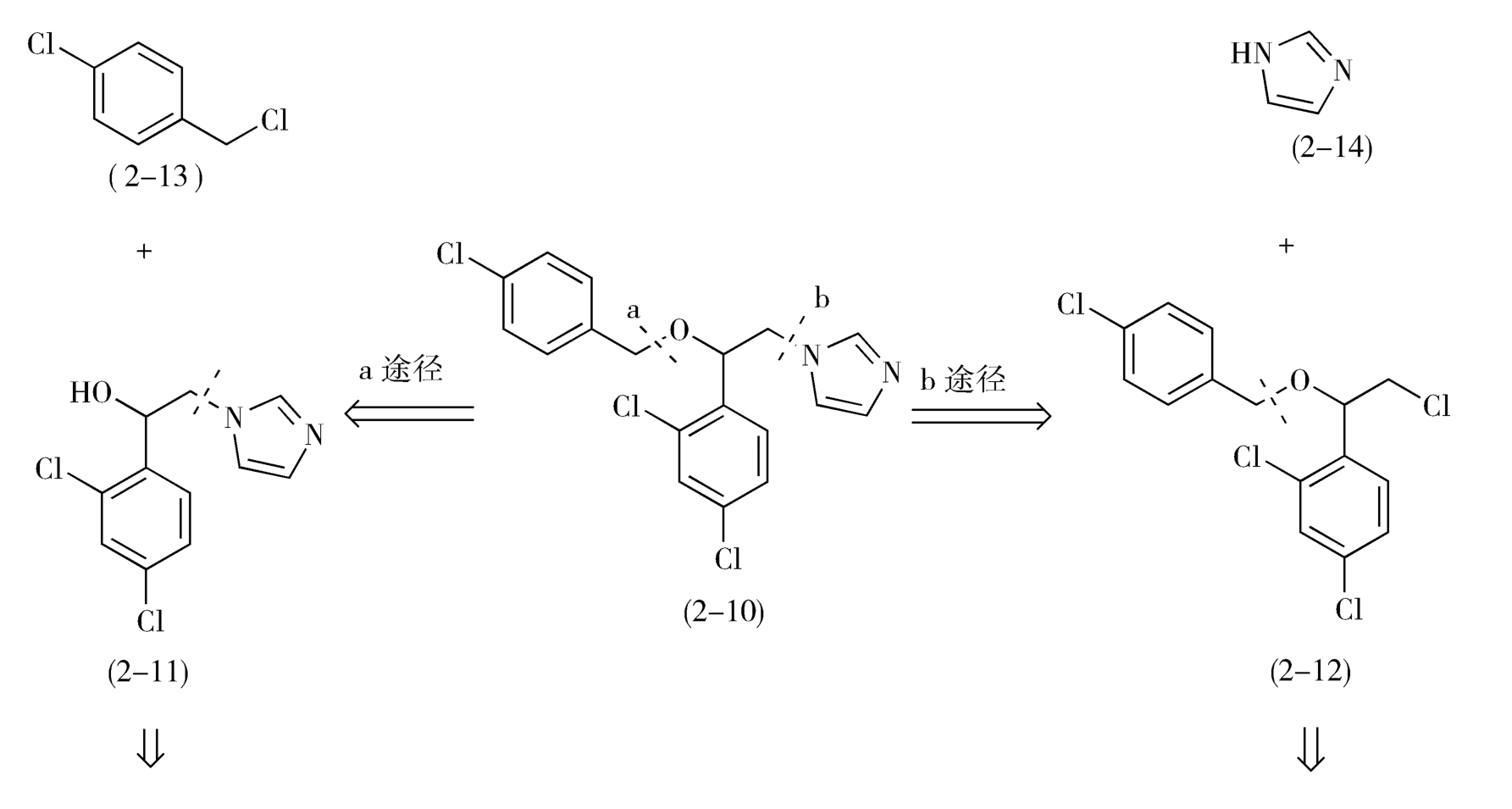
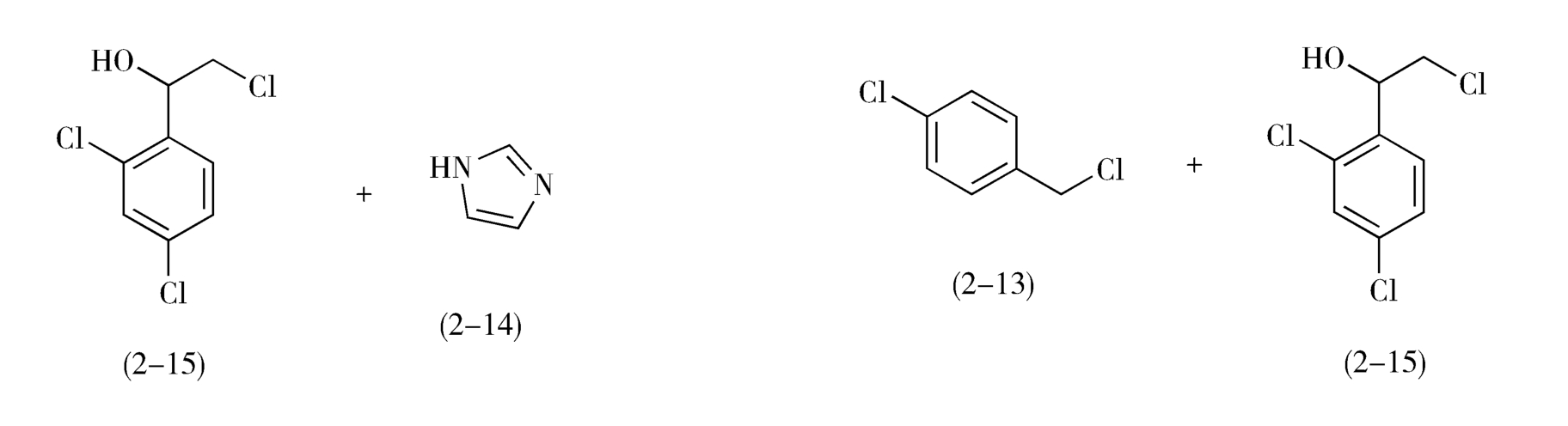
图2-7 益康唑的逆合成分析
如图2-8所示,1-(2,4-二氯苯基)-2-氯代乙醇(2-15)是一个仲醇,可由相应的酮还原制得,而其前体α-氯代-2,4-二氯苯乙酮(2-16)可由2,4-二氯苯(2-17)与氯乙酰氯(2-18)经Friedel-Crafts反应制备。益康唑的合成如下(图2-9)。
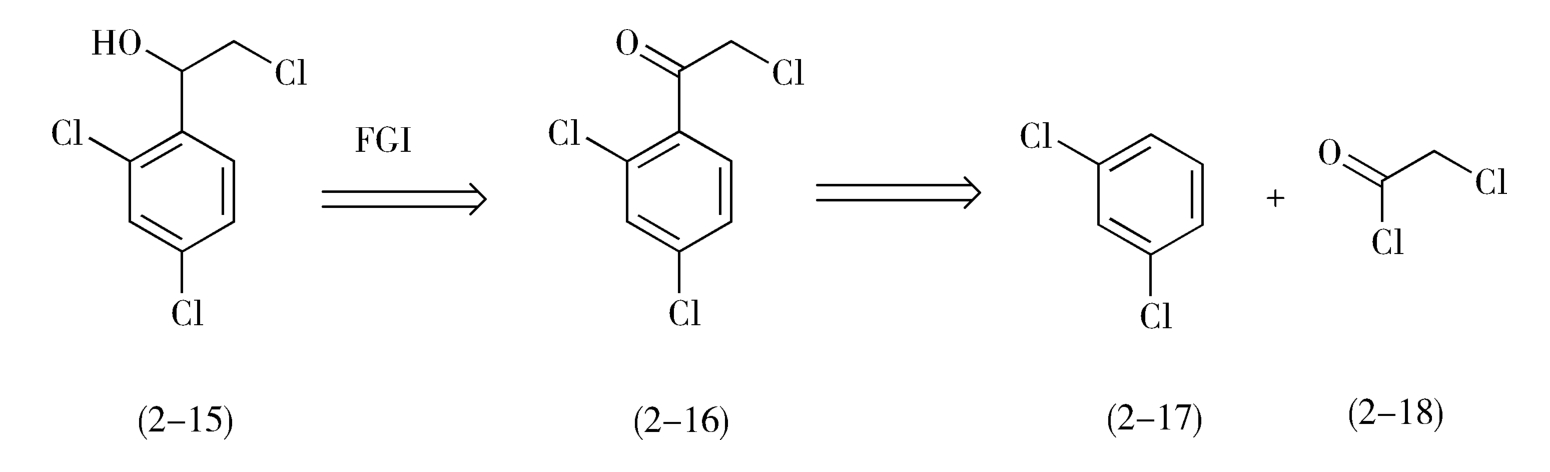
图2-8 1-(2,4-二氯苯基)-2-氯代乙醇(2-15)的逆合成分析
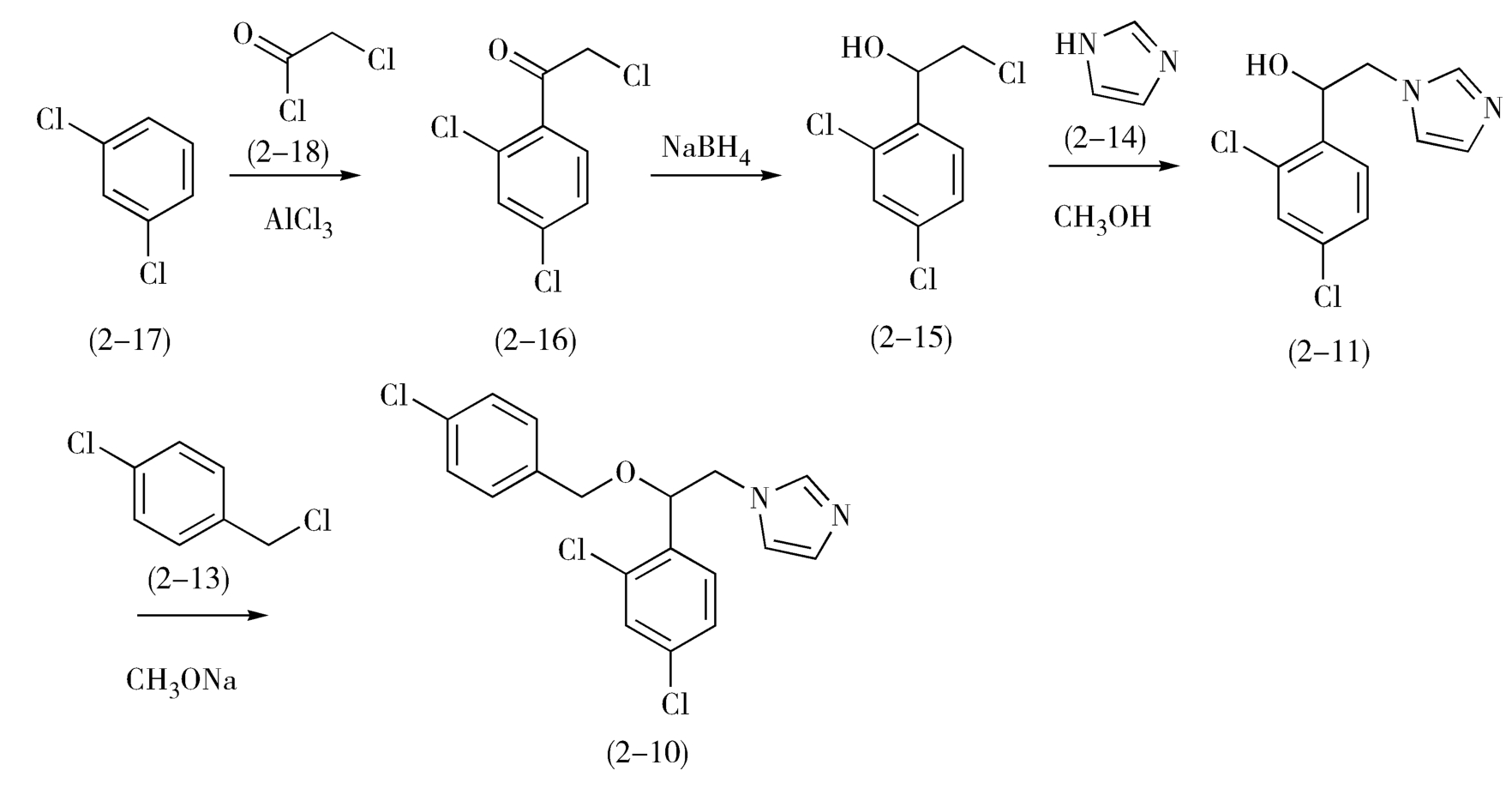
图2-9 益康唑的合成
2.从分子中间分拆 一般来说,碳-杂键易于合成,在分拆过程中处于优先考虑的地位。但是有时候首先分拆碳-碳键可简化合成过程,提高目标药物分子的合成收率。例如,中枢神经镇痛药哌替啶(pethidine,2-19)是含叔胺的脂环药物,在其分子分拆中,首先经过两次官能团的转化,然后从分子环键结合处分拆C—C键,再经过一次官能团转化和分拆C—N键,即可得到起始原料环氧乙烷和甲胺(图2-10)。
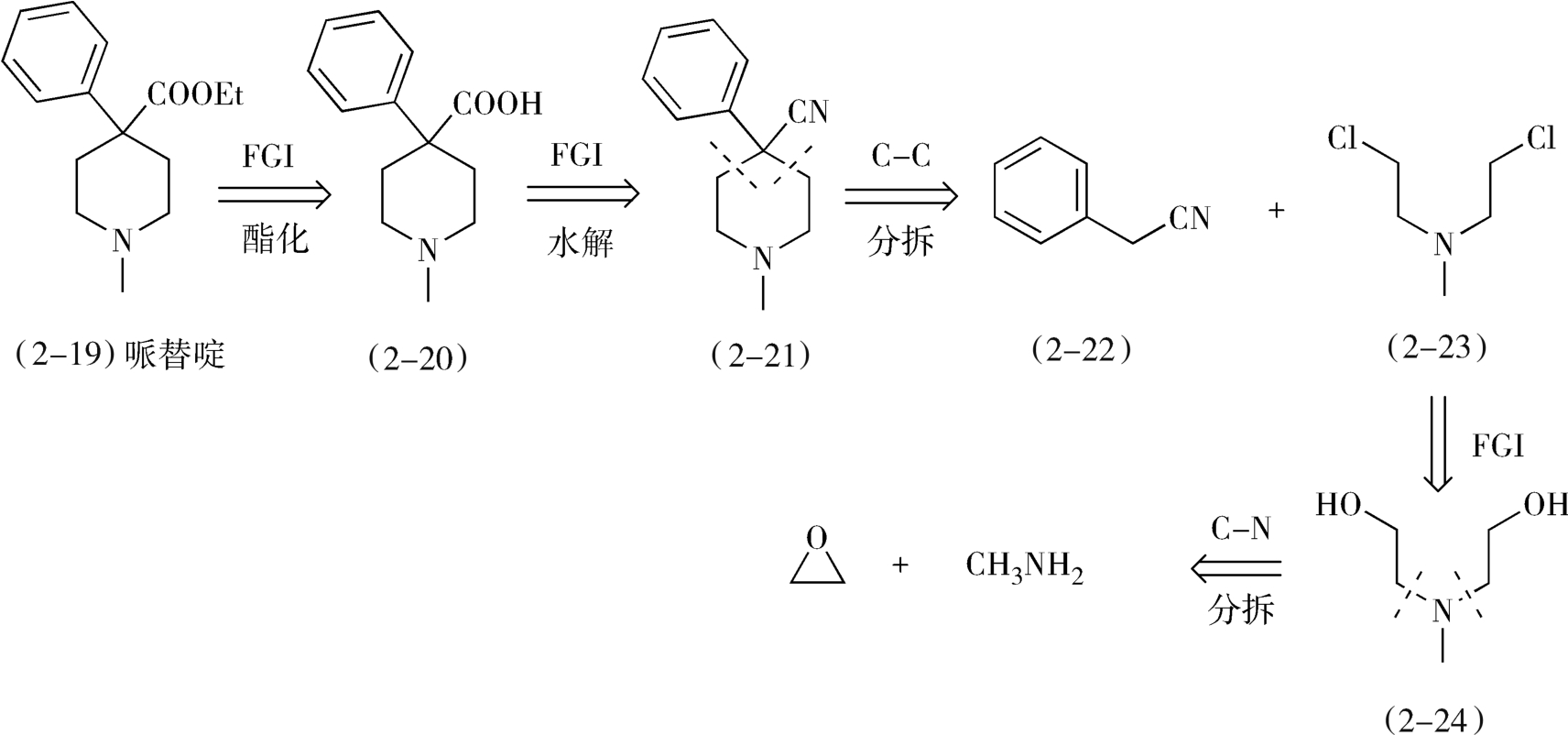
图2-10 哌替啶的逆合成分析
在本品的合成过程中,首先是使用环氧乙烷和甲胺发生氮烷基化反应,生成二(β-羟乙基)甲胺(2-24),然后使用氯化亚砜氯化2-24,生成二(β-氯乙基)甲胺(2-23),2-23与活性亚甲基化合物苯乙腈(2-22)在碱性条件(胺基钠)下缩合关脂肪环得到4-苯基-4-氰基哌啶(2-21),2-21在酸性条件下水解、酯化生成哌替啶(2-19)。
从这个例子中可以看到,中间体2-23和2-24均有对称性,因此需要考虑一步合成,从而简化合成步骤(可见,分子考察并不止于最终的目标化合物,而是包含在逆合成分析的全过程中)。
哌替啶的合成如图2-11。
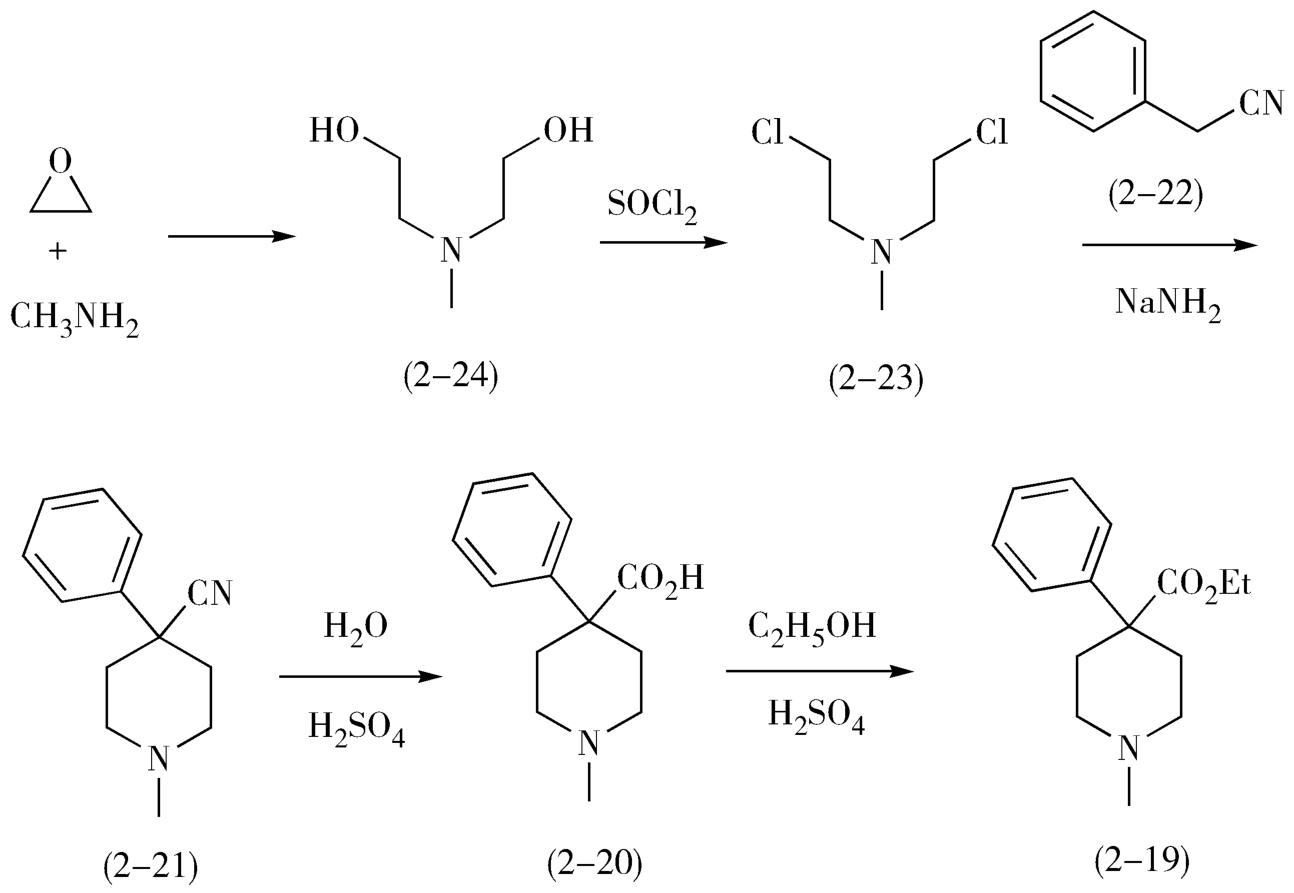
图2-11 哌替啶的合成
不论碳-碳键还是碳-杂键,从合成角度考虑逆合成转化顺序,特别是对一些比较复杂的药物分子,应着重强调从分子的中部分拆以获得汇聚法的合成;从分子中环键结合处或从分子的交叉点进行分拆。
例如,1998年上市的非甾体抗炎药环氧化酶-2选择性抑制剂西来曲葆(celebrex,2-25)的逆合成路线,首先选择在位于分子中部的吡唑处分拆分子,形成二酮(2-26)与4-磺酸氨基苯肼盐酸盐(2-27),二酮(2-26)可由4-甲基苯乙酮(2-28)和三氟乙酸乙酯(2-29)通过Claisen缩合反应制得(图2-12)。
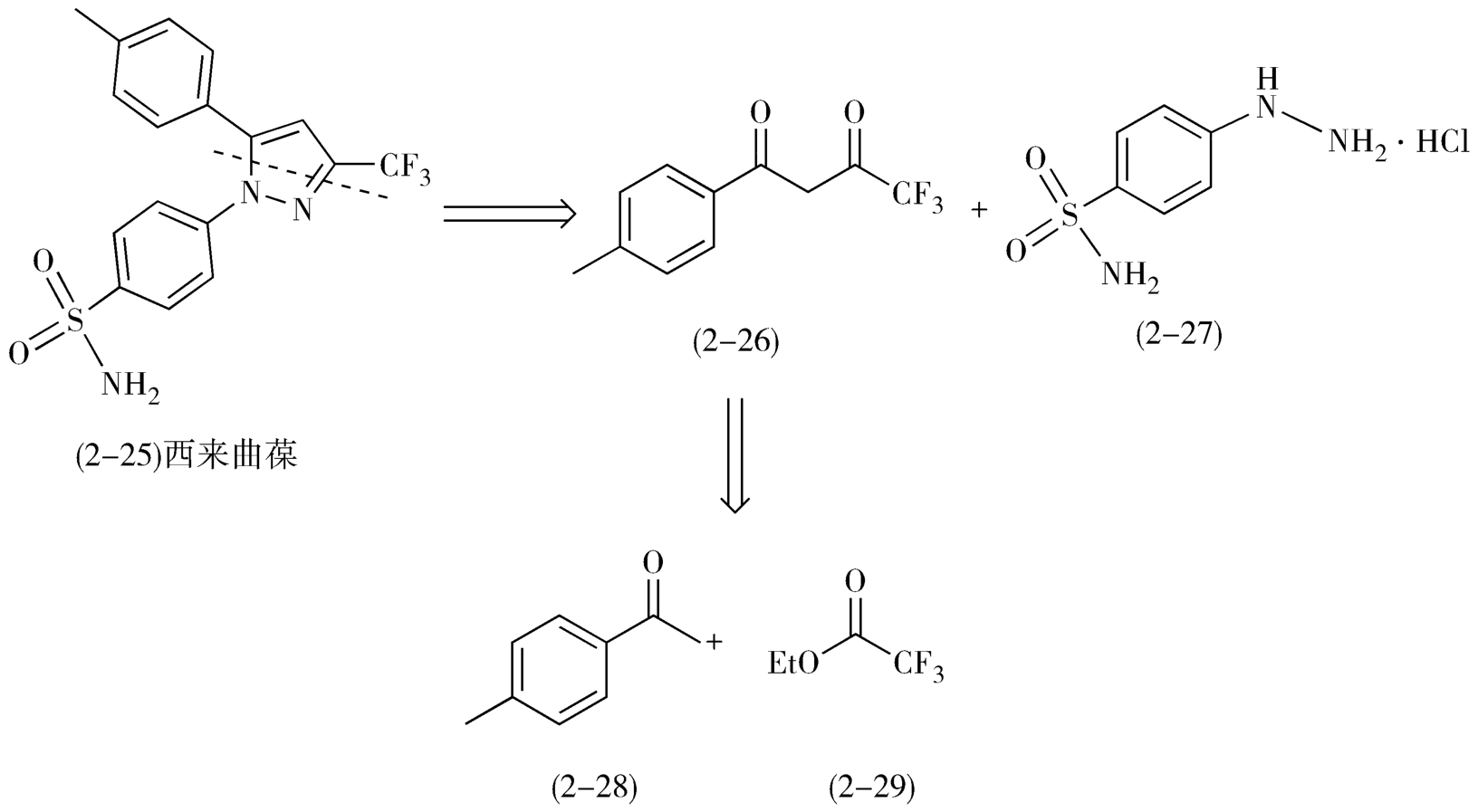
图2-12 西来曲葆的逆合成分析
将4-甲基苯乙酮(2-28)和三氟乙酸乙酯(2-29)在甲醇钠存在下、甲醇中回流,分子间缩合制备二酮2-26,2-26与4-磺酸氨基苯肼盐酸盐(2-27),在乙醇中回流缩合即得目标分子西来曲葆(2-25)。西来曲葆的合成如图2-13。
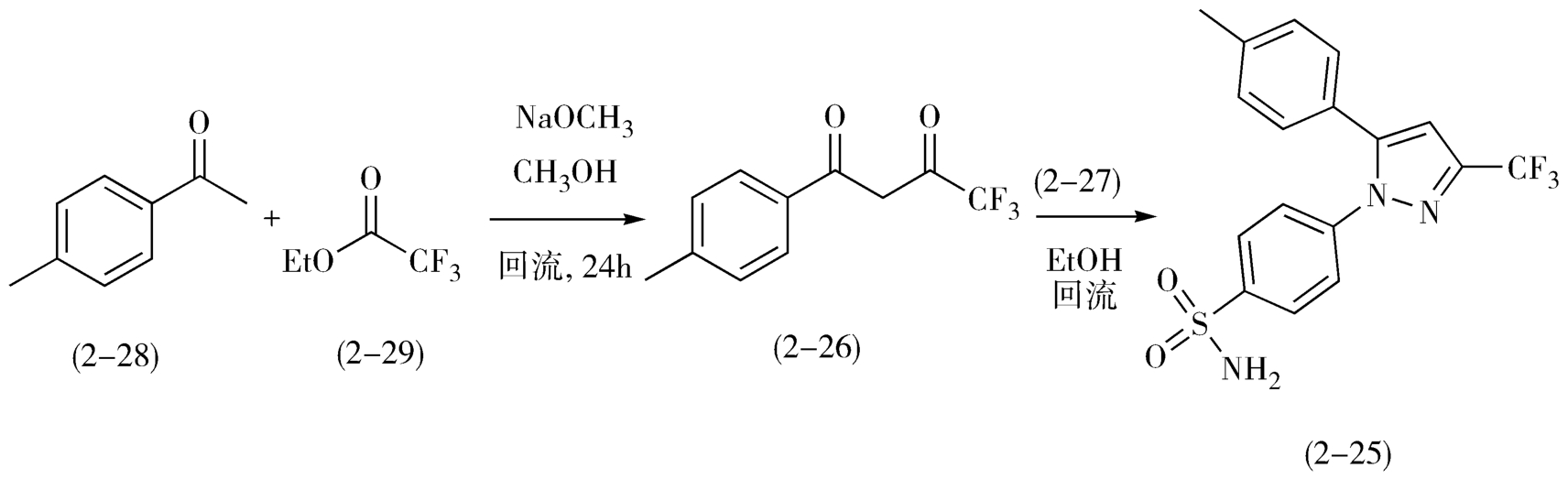
图2-13 西来曲葆的合成
从合成角度考虑逆合成转化顺序,还应注意首先安排相应反应产率高的转化,或相应反应成功把握大的转化,这是因为越到合成工作的最后环节,原料越为珍贵,失败的代价越为高昂,因此要尽一切可能增加成功率。要做到这一点需要对有机反应有切实深入的理解。
3.多键分拆 一个目标药物分子往往有多种分拆方法,分拆方法不同导致所应用的合成反应不同、合成路线的长短不同、反应条件不同,原辅料和产率也有所差别。因此可以尝试从合成反应优化合成转化顺序。
从合成反应优化合成转化顺序首先可以寻求多键分拆的策略:通过一步合成反应同时建立多个化学键是简化合成步骤的有效方法。上文谈到的哌替啶和西来曲葆的合成实际上都应用了这个策略。在设计降血脂他汀类药物美伐他汀(mevastatin,2-30)的逆合成路线时也应用了基于协同反应的多键分拆策略(图2-14)。
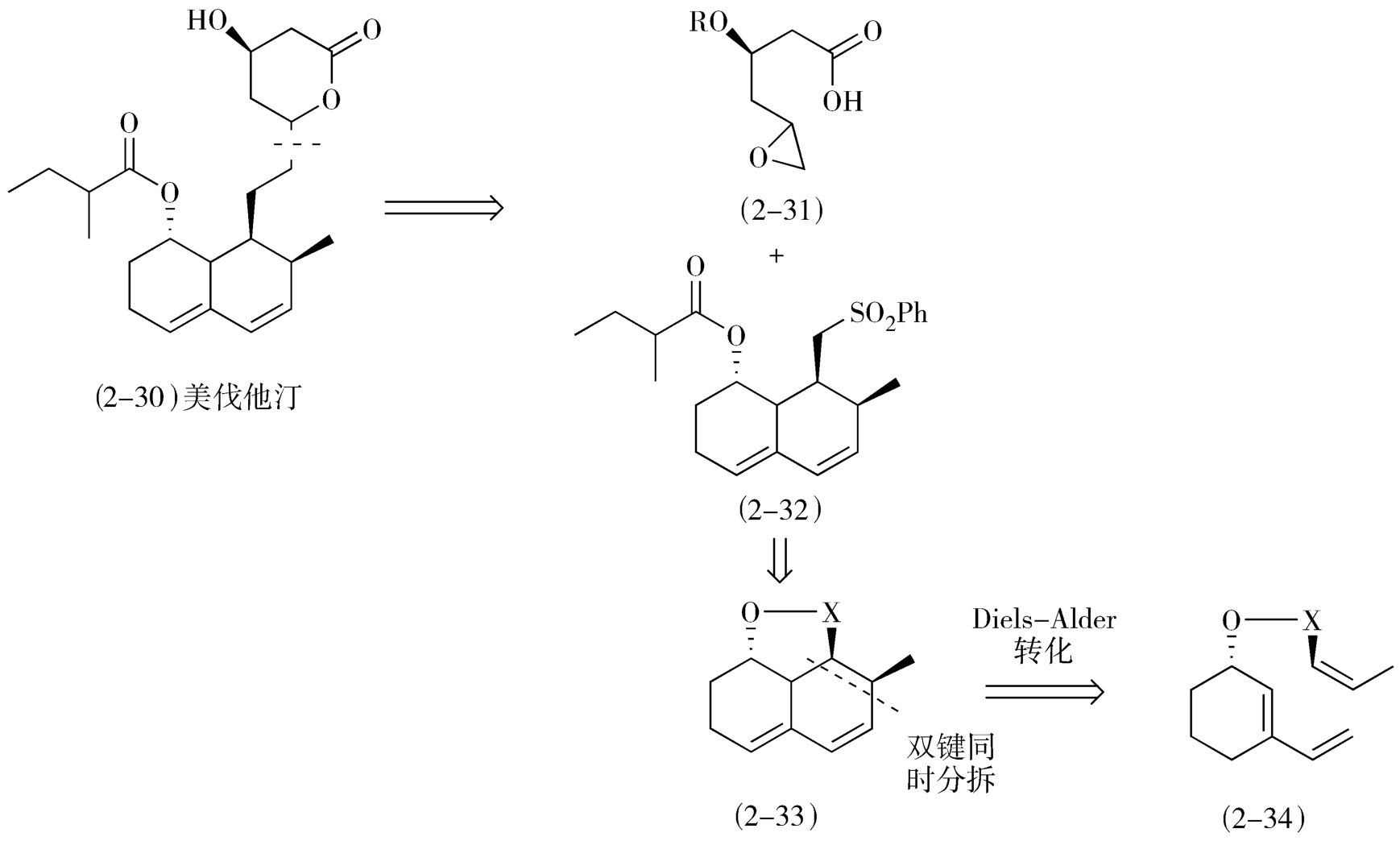
图2-14 美伐他汀的逆合成路线
Johnson甾体合成法利用了含氧基团电性效应引发的仿生-烯烃多重环合反应,可在此反应中同时建立三个碳-碳键和三个脂环,是合成甾体药物的理想方法(图2-15)。
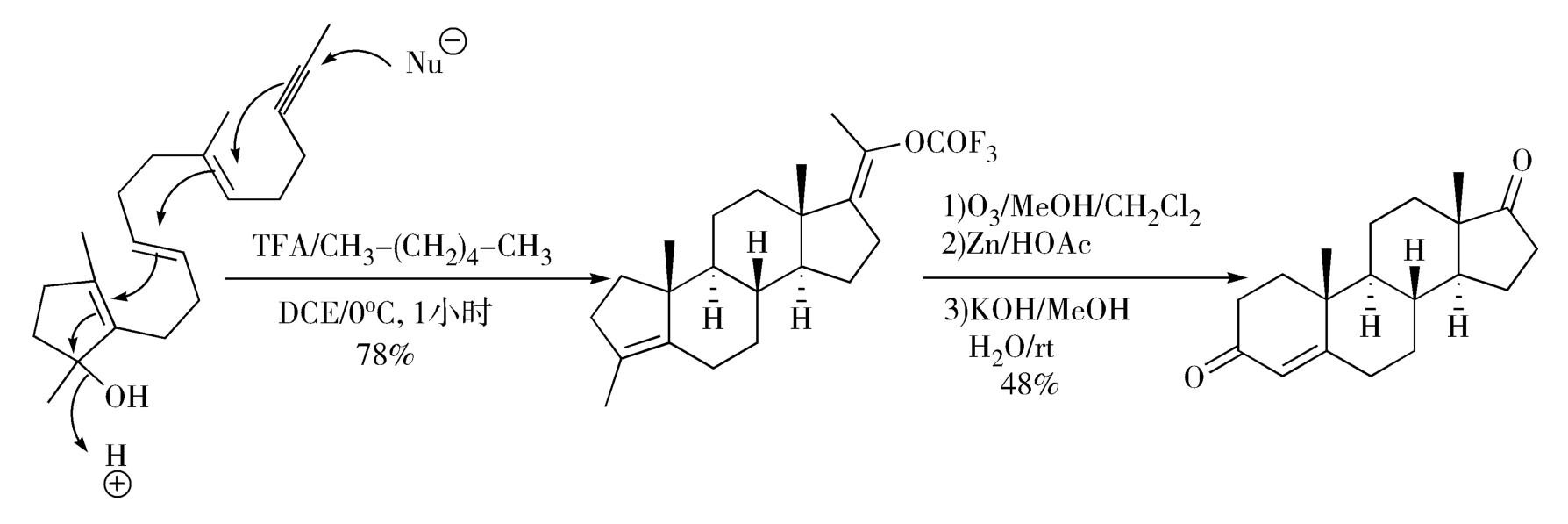
图2-15 Johnson甾体合成法
4.尝试联接与重排
可以尝试使用重排法和联接法。尽管分拆策略是逆合成分析的主要方法,但是利用联接与重排的方法往往可以简化合成路线。例如,苯并二氮
 类镇静催眠药氯氮
(chlordiazepoxide,2-38)的合成就是利用了2-氯甲基-4-苯基-6-氯-喹唑啉-3-氧化物(2-35)的扩环重排获得的(图2-16)。
类镇静催眠药氯氮
(chlordiazepoxide,2-38)的合成就是利用了2-氯甲基-4-苯基-6-氯-喹唑啉-3-氧化物(2-35)的扩环重排获得的(图2-16)。
联接法也是十分有用的逆合成分析方法,如经典的甾体全合成中D环(2-39)的形成,是将带有甲基酮侧链的D环推导得醛酮的中间体(2-40),而此中间体可用连接法推导得到其前体甲基环己烯(2-41),这里依据的是臭氧氧化反应(图2-17)。
由于各种原因,按照有机合成基本原理设计的合成路线在实际执行过程中常常会遇到一些始料未及的困难,因此在合成设计时一定要留有机动灵活的余地,最理想的情况是为每一个中间体的合成都准备两至三套方案。
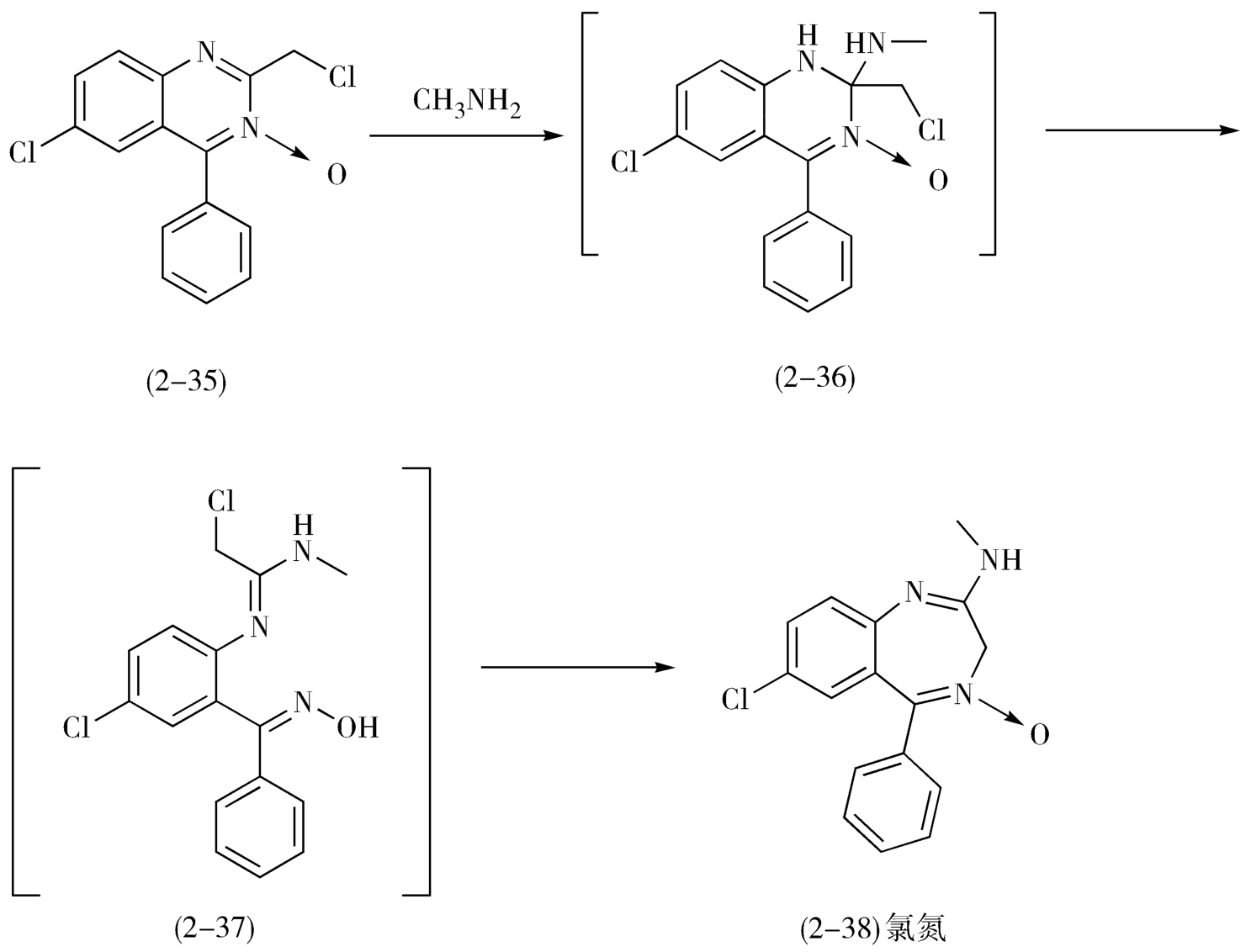
图2-16 氯氮
的合成
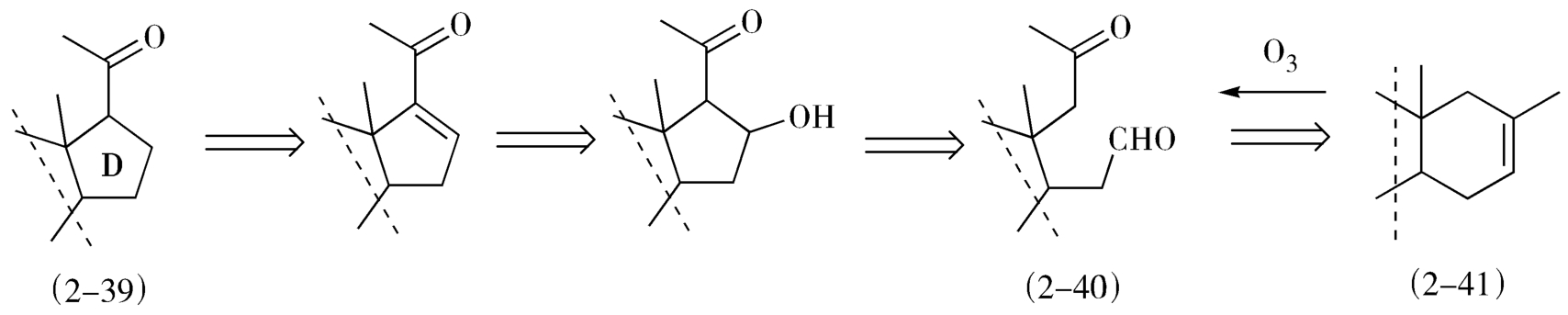
图2-17 甾体环D环的逆合成分析
总结以上论述,在逆合成分析中应考虑以下几条原则:
1.合成步骤尽量简短,这是评价一条合成路线好坏的关键考量之一。
2.切断化学键时只能使用已知并成熟的化学反应,就是说每一步反应都有切实可行的理论根据。
3.首先考虑切断碳-杂键,根据分子中的官能团,必须切断碳-碳键时如有可能,应力求最大简化,在分子中央切断,或在支化点上切断,或将环从链上切下来,如分子有对称性应予以利用。
4.所选择的切断或转化应该是相当于产率最高的反应,而且要尽量避免生成数种异构体的反应,除非已知这些异构体容易分离,其副产物有用途,能找到销路。
5.通过切断与转化,一直进行到可被接受的起始原料或易于制备的化合物为止。
6.重视联接与重排方法的合理利用。
在进行更有效率的逆合成分析时,还必须考虑目标化合物结构特点对选择分析步骤的影响,比如目标分子及中间体中官能团的排列与官能团互换的影响;与工业原料关系密切的特殊结构片段的存在(如苯环上羧基邻位有羟基时就要考虑原料选用水杨酸的可能性等);立体化学构型的影响;在目标分子或中间体中对称性的影响;在多环结构中“关键”键的存在。以上这些都是要使合成步骤尽量简短,因为每多一步反应就意味着产率的进一步降低,如果每一步的产率相近,自然是步骤越少总收率越高(详见第六章),但限于篇幅,这里不再一一说明。
逆合成分析工作结束后,将以上分析得到的起始原料与目标分子及中间经过的各个中间体按相反顺序用箭头连接起来,加入每一步反应条件就成为该药物合成的初步设计方案。然后进行实验以完善每一步操作,必要时进行部分修改,就成为一个完整的合成工艺设计;再经过中试放大,就可投入批量生产。整个研制过程至此才算真正结束。