
下载掌阅APP,畅读海量书库
立即打开



为了符合正交度原则,串联的两根GC柱必须使用不同且独立的分离机理。大部分情况下,GC×GC系统使用的第一根色谱柱为非极性固定相,如聚二甲基硅氧烷等,而第二根色谱柱的固定相极性较强,如聚乙二醇、聚苯基甲基硅氧烷或环糊精等。在这种设置下, 1 D上分析物通常依据沸点的高低被分离,而 2 D的分离常在恒温条件下进行,由分析物活度系数决定。当升温速率为3~10℃/min时, 2 D分析时间为几秒钟。因此,分析物通过两个独立的分离机理进行分离,形成正交。如果第一根色谱柱为非极性固定相,并与极性更强的第二根色谱柱相连,此设置通常称为正正交。而在反正交中, 1 D为中等极性或极性色谱柱, 2 D为非极性色谱柱。根据Adahchour等的统计,截至2005年,所有GC×GC文献中,使用正正交,即 1 D为非极性色谱柱, 2 D为中等极性色谱柱的文献约占80%。
GC×GC系统正交度越高,两根色谱柱的保留机理之间越独立,理论上来说化合物的分离效率就越高。但是,对于GC×GC而言,另外一个重要的作用是获得结构化的色谱图,即在色谱图中化合物按照类别分布,所有同一类别的化合物分布在一起。实际上,结构化的色谱图是GC×GC非常重要的一个作用,特别是在石油化学中。但是,对于痕量水平的环境分析,其主要目的是将目标分析物从潜在的基质干扰中分离出来,而结构化居于次要地位。尽管如此,正交度和结构化色谱图本身并不是分析的目标,而分离才是。根据Giddings所述,只要分离的维度和样品的维度完全匹配,结构化的色谱图完全可在非正交条件下获得。
Schoenmakers等认为在进行化合物的种类鉴别时,结构化这一现象是非常有效的手段。结构化的色谱图可产生样品的指纹图谱。正如前文所述,在鉴别石油样品和筛查环境样品中的持久性有机污染物(POPs)如多氯联苯(PCBs)时,由于这些类别的化合物包含大量的同系物,因此色谱图呈现结构化现象是非常有利的,如图3-1所示。图中,虚线所连接的为分子中含有相同氯取代基数目的C8同类物,同一组中每个C8同类物的位置由联苯骨架上的取代形式决定。
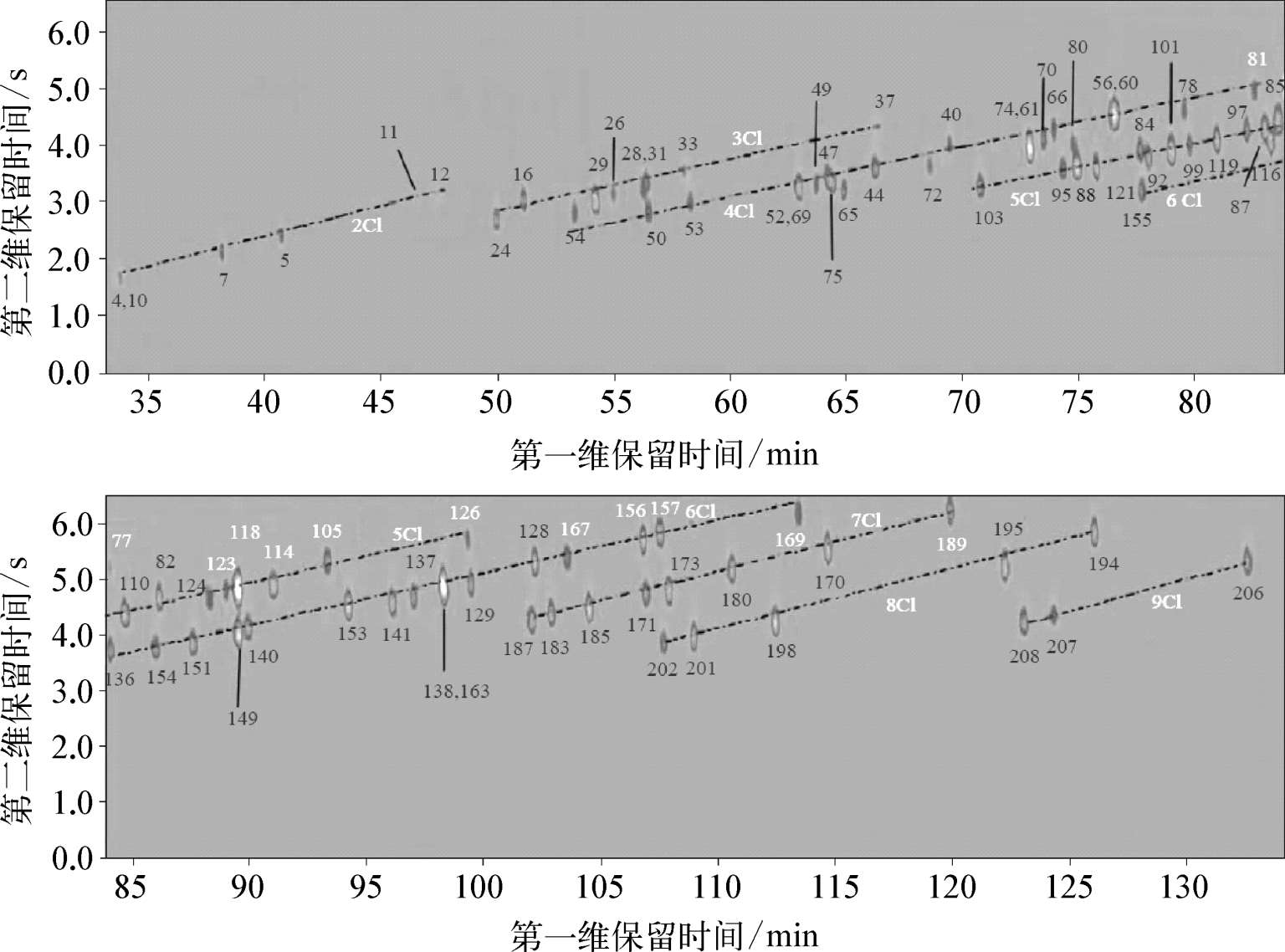
图3-1 90个PCBs混合物的GC×GC-μECD色谱图(HP-1×HT8)
Focant等使用了一个并不完全正交的GC×GC色谱柱系统,PCBs的分离仍呈现高度结构化,可实现基于同系物邻氯代程度的污染物的识别。
如上文所述,正交度带来了许多优点,部分研究证明反正交方法,即 1 D为极性色谱柱, 2 D为非极性色谱柱,和正正交具有互补性。例如,图3-2为同一个橄榄油提取物样品的两个GC×GC色谱图。图3-2A中,正正交方法对于极性相对较低的分析物产生了较好的分离结果。在 2 D中,这些分析物与其他组分相比保留较弱,在色谱图 2 D中为0.3~1.7 s以带状形式呈现。在大部分情况下都可以从极性基质中获得有效的分离。但是,图3-2A中与极性化合物的共馏出出现了峰迂回现象。如 1 D中20~25 min出现了细长的带状的点,这是一个显著的缺陷。反正交方法,从另一个方面来说,更适合分离极性较强的分析物。由于极性较强的化合物在 1 D色谱柱上保留很强,大多数情况下都可以与非极性组分分离,如图3-2B的 1 D中15~33 min, 2 D中1.0~2.6 s的区域。与正正交方法相比,对于极性较强的化合物不存在峰迂回问题,且峰形较好,而对于非极性分析物则存在峰迂回问题。
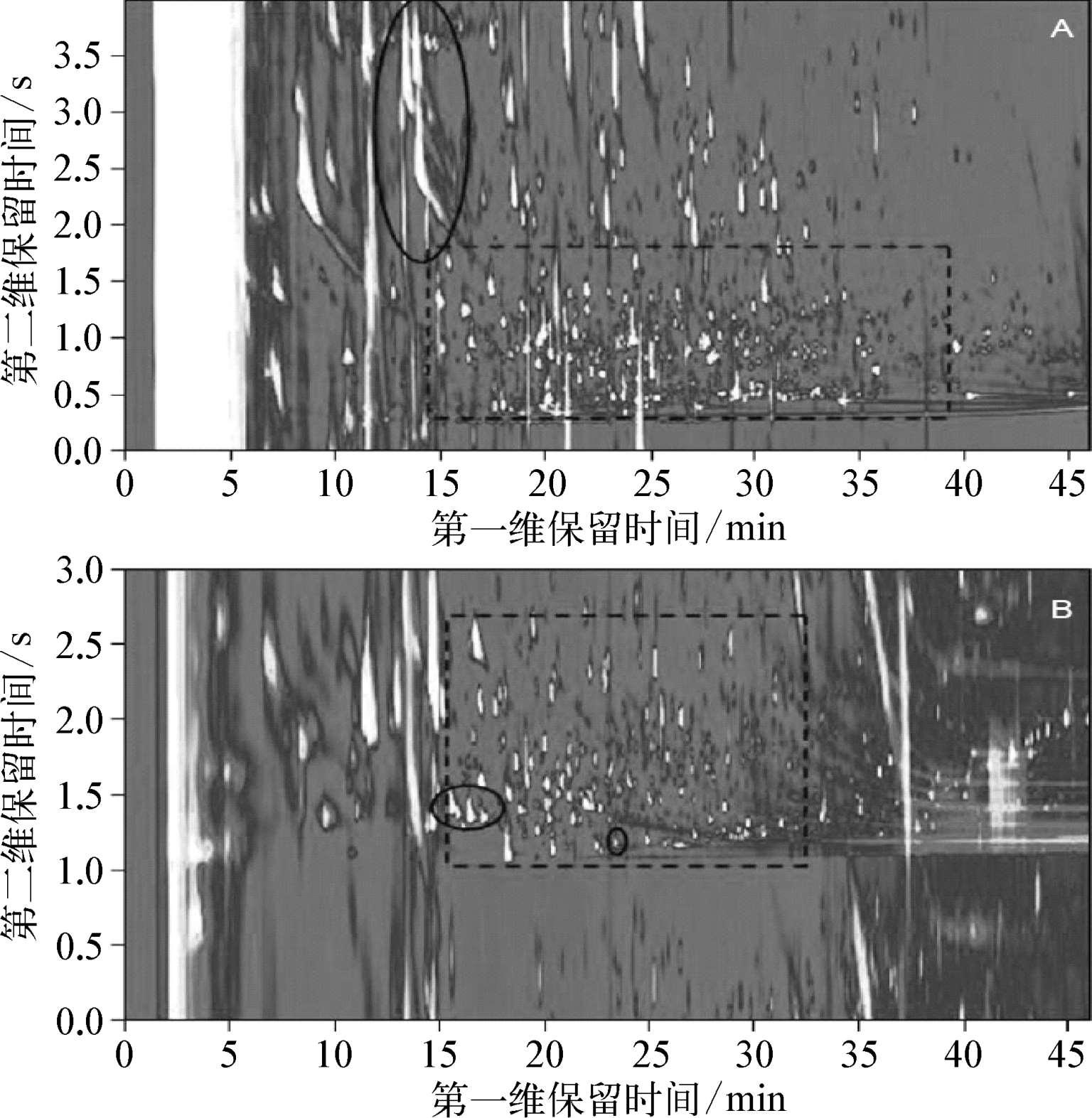
图3-2 橄榄油提取物的GC×GC-FID色谱图
(A)使用正正交方法,(B)使用反正交方法。三处圆圈为3 -甲基丁酸和三个醇,1 -己醇,顺式 3 -己烯醇和反式 2 -己烯醇。
(A)中虚线区域主要为非极性组分,(B)中虚线区域主要为极性组分。
在GC×GC中, 1 D的色谱柱通常为15~60 m长,内径在0.25~0.53 mm,膜厚为0.25~1μm,所产生的色谱峰峰宽一般为5~30 s,与传统1DGC常用的色谱柱并无区别。最主要是为了保证在共馏出的分析物进入 2 D分离前,进入调制器的色谱峰有足够大的峰宽,能够符合调制器取样次数的要求。
理想状况下,为了避免产生峰迂回效应,脉冲出调制器的化合物的 2 D保留时间( 2 t R )应该小于或最多等于 P M ,即一个完整的调制循环的时间。当化合物的 2 t R 超过GC×GC系统的 P M ,就会导致这个化合物在下一个或其后的调制中出现,从而产生峰迂回现象。为了使峰迂回效应最小化, 2 D色谱柱通常较短,一般为0.5~1.5 m长,且内径比 1 D色谱柱要小,膜厚也通常降低至0.1~0.25μm范围内,以提升分离效率。一般情况下,与内径较大的色谱柱相比,内径较小的色谱柱可提供较高的峰容量。但是在GC×GC中,如果使用一根细孔径的色谱柱作为 2 D,在 2 D色谱柱中的压力要比使用一根内径较大的色谱柱作为 2 D要高得多,这就会导致 1 D色谱柱的色谱过程减缓,但可以使用一根较长、内径较宽的色谱柱作为 2 D来解决。此外,还可以将 2 D色谱柱放在另外一个柱温箱里以进行温度的独立控制,来避免峰迂回的产生。虽然峰迂回常被看作优化不完全或者分离不理想的象征,但是,只要化合物的峰迂回不产生新的共馏出现象,没有必要刻意花费过多的时间和精力来避免峰迂回。即使出现了峰迂回,分析人员可以重新建立更为清晰的色谱图。峰迂回最主要的缺点是导致 2 D色谱峰略微变宽,并可能最终导致峰容量降低。另一个缺点是当使用单维检测器时,可能导致峰重叠或共馏出现象。
目前,大部分GC×GC的应用都是基于传统1DGC方法发展而来,只是在GC×GC中需要进行一系列的优化,如色谱柱系统、载气流速和升温速率等参数。Beens等建立了一个基于Excel的计算器,对于特定的一组色谱柱,可进行最佳分离条件的预测。这一模型显示使用内径为 0.1 mm的色谱柱作为 2 D可能并不是分离的最优条件。在他们的研究中,使用内径为 0.25~0.32 mm的色谱柱作为 1 D,与内径为0.15~0.18 mm的 2 D色谱柱共同使用,得到了很好的分离结果。由于扩散与色谱柱内压力相关,当 2 D色谱柱内径较小时,其流速应比1DGC中使用的流速小得多。因此,一般情况下, 1 D色谱柱最佳载气流速应该为1DGC流速的一半左右,而GC×GC的总分析时间应该为1DGC分析时间的两倍。
大部分GC×GC领域的研究都希望在保持较好灵敏度的基础上获得一定的分离选择性,然而,这在一定程度上会导致分析时间过长。Harynuk和Marriott提出另外一种优化方法,为了分析速度而牺牲部分选择性,因此在 1 D中使用了一根快速的GC色谱柱。他们将一根BPX 5 色谱柱(5 m ×0.25 mm×0.25μm)与一根BPX 50色谱柱(0.3 m×0.15 mm×0.15μm)相连,化合物的馏出温度大幅下降,从而使得分子量相对较高的化合物能够在较短时间内完成分析。
Zhu等在 1 D和 2 D分别使用了不同膜厚的色谱柱进行组合和测试。他们得出结论,当在 2 D中不要求分辨率高而要求速度快时,在 1 D中使用液膜较薄的色谱柱,在 2 D中使用内径较窄的色谱柱更为合适。相反,如果分辨率更为重要时,可以在 1 D使用膜厚为 0.25~0.5μm的色谱柱,在 2 D使用宽内径(150μm)的色谱柱。
在GC×GC柱组合中,有一个特殊的组合是使用互相平行的两根色谱柱而不是单一的一根色谱柱作为 2 D,通过将聚焦后的脉冲分开并传递到两根平行的 2 D色谱柱上来实现。这种组合由Seeley等首次报道提出,他们在调制阀后使用了一个馏出物分流器,用来将选定的片段发送到两根平行且不同的第二根色谱柱上。由此所产生的技术,称为双二维柱GC×GC(GC×2GC),在一次分析后可产生两个二维轮廓图或像素图。
任何已经在1DGC中使用的固定相都可以用于GC×GC,也可以根据目标分析物和固定相之间的相互作用来选择合适的固定相。虽然商业化细孔径色谱柱的数量在过去的二十几年内有了显著的增长,但是对于GC×GC中 2 D所要求的极性更强、热稳定性更好的色谱柱的选择仍然非常有限。这一问题的存在显著降低了GC×GC的应用范围和潜力。尽管如此,一些专注于生产客制化色谱柱的实验室已经开发了适用于GC×GC、特异性更强的固定相。Cordero等研究了混合固定相色谱柱的正交度和分离空间占有率。固定相为25%∶75%的聚乙二醇∶二甲基聚硅氧烷混合物的色谱柱用于 2 D的研究表明,这类色谱柱提高了天然挥发性化合物的分离度。此外,在 2 D使用中等极性固定相可以实现极性化合物的高效分析,主要体现在色谱峰较窄且峰形拖尾少。而极性更强的色谱柱,如聚乙二醇为固定相的情况,可能导致保留值混乱,从而影响定量的精密度。Sidisky等提出了一种极具应用前景的离子液体固定相,具有极佳的耐热性能(高达240℃)。Seeley等也在GC×GC分离中使用了一种高温磷离子液体色谱柱,实验结果证明这些固定相都是GC×GC中 2 D色谱柱很好的选择。
在 1 D采用传统色谱柱,使用近似1DGC的载气流速,使得所有的进样技术都可以直接运用于GC×GC,例如,分流、不分流、大体积、程序升温(PTV)和固相微萃取(SPME)进样等。进一步的,任何基于传统GC分离的方法均可用于GC×GC,而不需要对已有的进样技术进行改进或优化,因此本书不再就这些问题进行阐述。