
下载掌阅APP,畅读海量书库
立即打开



离解吸附是一种通过表面的反应,但它不一定会导致表面原子的去除或刻蚀。为了进行刻蚀,表面上的原子必须重新排列成包含至少一个表面原子的分子。这些新形成的分子必须以比吸附的分子或原子本身更低的能量解吸。例如,F 2 可以物理吸附在硅表面,并分解成化学吸附的F原子,从而形成SiF x 。SiF x 与硅的键比F弱。因此,在该系统中,加热含氟硅样品可去除SiF 2 和SiF 4 (Engstrom等人,1988)。
在图2.5中,反应坐标从系统化学吸附原子/表面到反应产物/表面的转变由箭头表示。在最简单的情况下,这只是对键表示形式的改变。实际上,这种转变可能涉及化学反应,以重新排列键并形成新的分子。在这种情况下,解吸坐标之间的转变涉及具有自身反应坐标和能量势垒的化学反应。如果反应的能量势垒高于解吸势垒,则该反应将是速率限制步骤。
从先前氟刻蚀的硅表面去除氟而不损失任何额外的硅原子是一项挑战。在这种情况下,必须使用另一种表面反应,例如与水蒸气的反应,以除去HF形式的氟。涉及两个或更多分子或原子的多重表面反应也是热ALE的机理,将在第4.1节中介绍。
通过计算吉布斯自由能Δ G °,可以预测表面反应进行的可能性:

式中,Δ H °是焓的变化,表示反应热;Δ S °是反应的熵变化,反映了系统有序性的变化;上标表示标准温度和压力,分别为273.15K和10 5 Pa。化学反应的吉布斯自由能可以使用商用软件程序计算。
当式(2.4)中Δ G °为负值时,则该反应在热力学上是有利的,并且称为自发的。需要自发反应来实现热刻蚀和热ALE。因此,热力学计算是热刻蚀反应可行性的第一次检查。Δ H °和Δ S °的单个值为刻蚀反应的温度行为提供指导。负Δ H °意味着反应是放热的。平衡将发生变化,有利于在较低的温度下进行刻蚀。如果Δ S °是负的,也更有利于反应在较低温度下进行。即使刻蚀反应在热力学上是有利的,反应动力学也可能阻碍有意义的刻蚀速率。反应路径可能包括必须克服的活化能势垒。这些势垒决定了反应速率。反应动力学的计算要复杂得多。
涉及一种以上吸附质的表面反应可根据其动力学进行分类,如图2.6所示。在Langmuir-Hinshelwood机制中,两个分子吸附在相邻的位置上,被吸附的分子发生反应。对于Eley-Rideal反应,只有一个分子吸附,另一个分子直接从气相中与之反应,例如,通过直接碰撞。
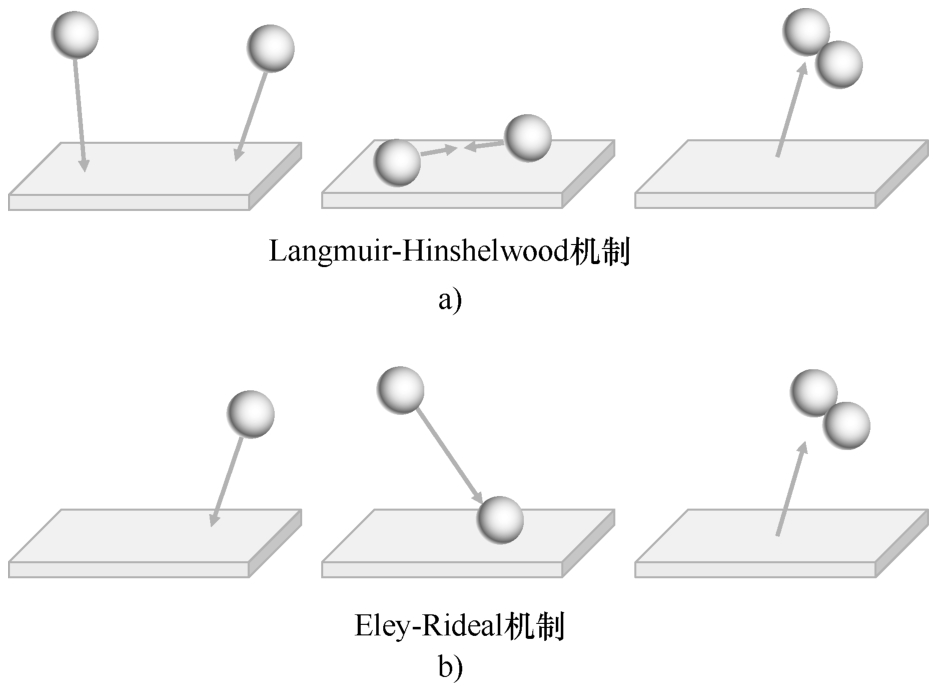
图2.6 Langmuir-Hinshelwood(图a)和Eley-Rideal(图b)表面反应机制示意图