
下载掌阅APP,畅读海量书库
立即打开



目前主要有两种形成液态金属纳米颗粒的方法,最简单的方法依赖于在液态金属表面上形成自限界面氧化层(图2-7),但是,天然氧化层不稳定,可溶解在酸或碱溶液中。一种更精准的表面钝化技术是利用物理吸附或者化学反应。物理吸附以静电吸附为主;化学反应则是利用在液态金属表面的配体。这种配体通常由结合到液态金属表面的锚定基团和可自组装成有序阻挡层的尾基团组成。常用的锚定基团包括硫醇基团,非硫醇锚定物通常会附着到界面氧化物而不是裸的液态金属或金属合金表面。
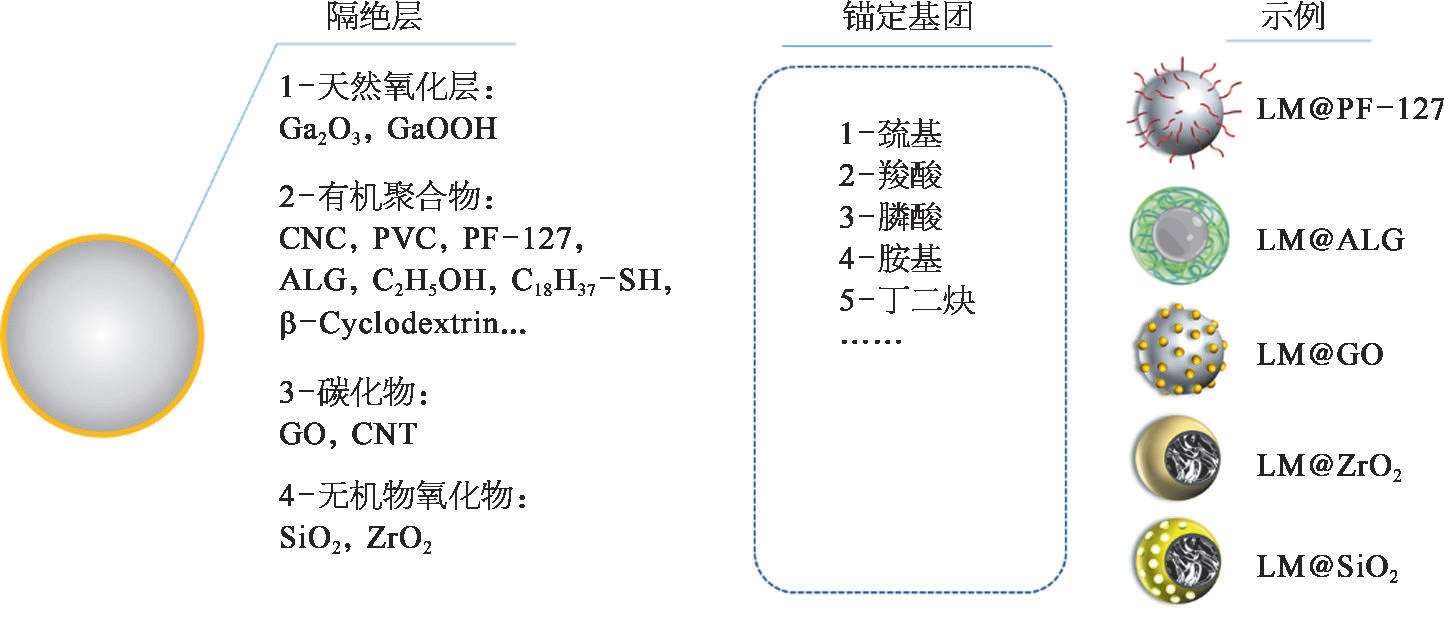
图2-7 液态金属纳米材料的表面修饰方法
利用有机配体对镓基液态金属纳米颗粒表面修饰是增强其分散性和功能性的常用策略,在有机配体作用下,镓基液态金属表面也会形成厚度不同的反应氧化层。Buonsanti团队利用显微成像和光谱表征技术深入探讨了有机配体和液态金属氧化层之间的相互作用 [34] ,通过实验证实胺和羧酸基团可以促进较厚氧化层的生成,而硫醇和膦基团则会阻碍氧化层的生长[图2-8(a)~(c)],并从热力学和反应动力学的角度进行了剖析[图2-8(d)]。从热力学角度看,有机配体对Ga或者Ga 2 O 3 的亲和倾向起主导作用;从纯动力学的角度来看,应考虑有机配体在液态金属表面的堆积密度,因为它可以改变氧气对金属表面位点的接触。对于油酸(OLAC)和油胺(OLAM),它们有相同的烷基链,只是基团上有所不同,因此二者在液态金属表面的堆积密度可以认为是相同的,不会对氧化物生长的表面位点造成差别。根据软硬酸碱理论,羧酸盐是比胺更硬的碱,因此具有对硬酸(Ga 3+ )更高的亲和力。OLAC不仅有利于Ga 3+ 的外向扩散,而且在氧化物形成过程中可更好地钝化氧化物,最终促进形成比OLAM更厚的氧化层。而对于三辛基膦(TOP)和十二硫醇(DDT),膦和硫醇是软碱,对软酸具有更大的亲和力,对于阻碍镓基纳米颗粒的氧化,它们是合适的配体。膦是比硫醇更软的碱,因此对金属位点具有更强的亲和力。除了软硬酸碱理论,TOP比DDT(0.8D)有更大的电偶极矩(>1.2D),因此它可能在更大程度上反对Mott电位,最终减少氧化厚度。总的来说,热力学表明DDT的氧化壳比TOP的厚。
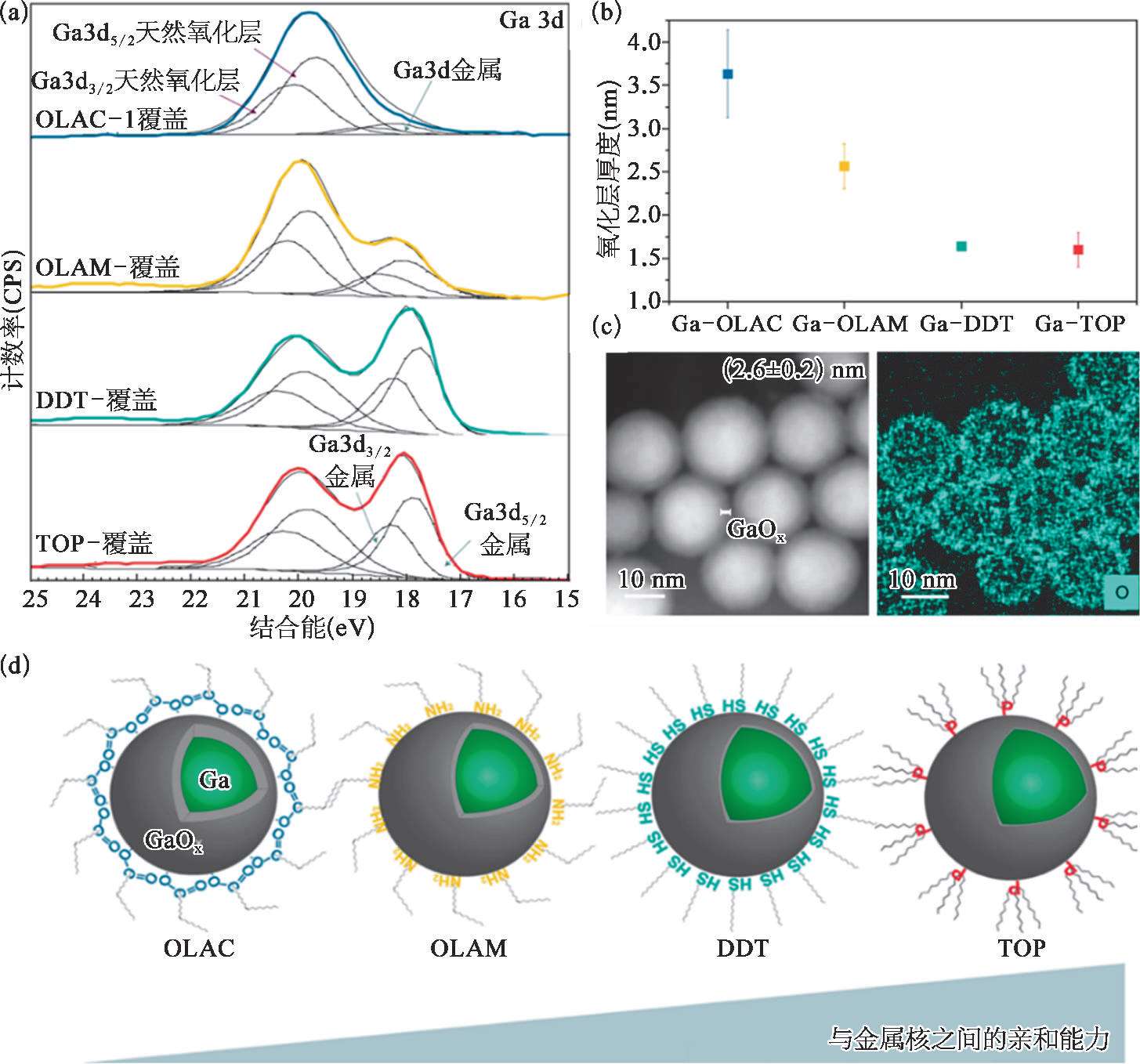
图2-8 表面修饰基团对液态金属纳米颗粒表面氧化层厚度的影响 [34]
(a)通过XPS光谱可以计算不同有机配体修饰的颗粒中Ga和Ga2O 3 的比例,以及氧化层的厚度;(b)不同配体对应的氧化层厚度OLAC>OLAM>DDT~TOP;(c)HAADF-STEM对氧化层厚度的表征说明了实际值和计算值接近;(d)从热力学和动力学的角度剖析。
形成阻挡层的典型材料包括有机化合物、碳基材料和无机氧化物材料等等,下面将作具体介绍。
利用有机化合物对液态金属纳米颗粒进行修饰是目前较为常用的表面修饰方法,已报道过的有机聚合物包括PVC、CNC、PF-127、ALG、C 2 H 5 OH、C 18 H 37 -SH、β-Cyclodextrin等,锚定基团包括巯基、羧基、膦酸基团、氨基、丁二炔等。在水溶液中液态金属微纳米颗粒表面的修饰物可大致分为4大类,即表面活性剂类、聚合物类、硫醇修饰的PEG类和生物分子类 [35] (表2-2)。
表2-2 常见的液态金属表面修饰物

1. 不同表面活性剂修饰的纳米液态金属颗粒
不同有机聚合物修饰对于纳米液态金属的形状、粒径、表面电势会产生不同的影响。通过扫描电子显微镜对CTAB、PEG-SH、SDS的液态金属微纳米颗粒的形貌进行观察,结果如图2-9所示,在37℃的去离子水溶液中放置48h后,CTAB修饰的纳米液态金属颗粒从球形变为棒状的GaOOH,而PEG-SH和SDS修饰的纳米液态金属颗粒仍为球形,其表面增加了较多不规则颗粒物,但没有产生整体性的棒状变形。此外,研究者还表征了3种溶液在4℃去离子水溶液环境中48h前后的微观形貌。由图2-9可知,CTAB修饰的纳米颗粒部分由球形变为米状,即产生了向棒状GaOOH转变的趋势;其余两种修饰物包裹的颗粒物则仍为球形。

图2-9 3种修饰物包裹的液态金属微纳米颗粒 [42]
(a)新鲜制备;(b)在4℃环境下48h颗粒微观形貌图。
2. 阳离子表面活性剂修饰制备可变形纳米液态金属颗粒
以阳离子表面活性剂CTAB修饰表面带负电的液态金属微纳米颗粒作为示例。CTAB不仅具有保持其稳定性的作用,还具有促进液态金属颗粒产生变形的功能。图2-10(a)展示了可变形液态金属微纳米颗粒的制备流程。将大块液态金属EGaIn放置于盛有CTAB溶液的烧杯中,通过大功率探头式超声的空化作用,将液态金属剪切为液态金属微纳米颗粒。在超声过程中,制备得到的颗粒为球形。通过将其置于去离子水中并加热不同的时间,即可得到不同形貌的微纳米颗粒。
图2-10(b)展示了扫描电镜下液态金属微纳米颗粒由球形向棒状转变的微观形貌。可以看出,在CTAB的修饰下,温度是促进液态金属从球形向棒状转变的重要影响因素。20℃时,球形液态金属转变为棒状需要24 h。随温度升高,所需要的棒状转变时间越短。50℃时,20 min液态金属即变为棒状,24h后则为棱角分明的长方体状。在球形液态金属向棒状转变时,可以发现在30℃放置20 min至8h时,出现了一种类米状形貌的液态金属颗粒,即米状液态金属。根据扫描电子显微镜下球形、米状及棒状液态金属微纳米颗粒的形貌,可归纳出形貌与加热处理时间与温度的关系图[图2-10(c)],为超声辅助加热法制备可变形多形貌的液态金属微纳米颗粒提供指导。
该方法能够制备具有可变形能力的液态金属微纳米颗粒,但也反映出当液态金属微纳米颗粒悬浮液置于20~30℃室温环境下时,其形貌会向棒状转变。通过冷冻干燥的方式将液态金属微纳米颗粒冻干为粉末,该方法通过冷冻使液态金属纳米颗粒溶液中的冰直接升华的方式,消除了由于水存在使液态金属产生持续变形的问题,可得到具有特定形状的颗粒。
图2-11(a)展示了所制备的液态金属微纳米颗粒的SEM和TEM形貌图。由图可知,该方法能够大批量制备球形、米状和棒状的可变形的液态金属微纳米颗粒,所制备的颗粒具有良好的形貌、较为均一的尺寸。为探究球形、米状和棒状液态金属微纳米颗粒的组成机理,通过XPS对三者的物质组成进行分析。如图2-11(b)所示,球形液态金属微纳米颗粒的主要组成为Ga和Ga 2 O 3 ,米状颗粒组成为Ga和Ga 2 O 3 及GaOOH,棒状的组成与米状类似,但GaOOH的峰高变高,峰面积变大,相对含量增多,间接表明了米状液态金属微纳米颗粒为球形的Ga向GaOOH转变的中间产物。所制备的棒状液态金属微纳米颗粒也非GaOOH,可能由于其未产生彻底的氧化,所观察到的棒状内部仍有未发生转化的Ga所致。
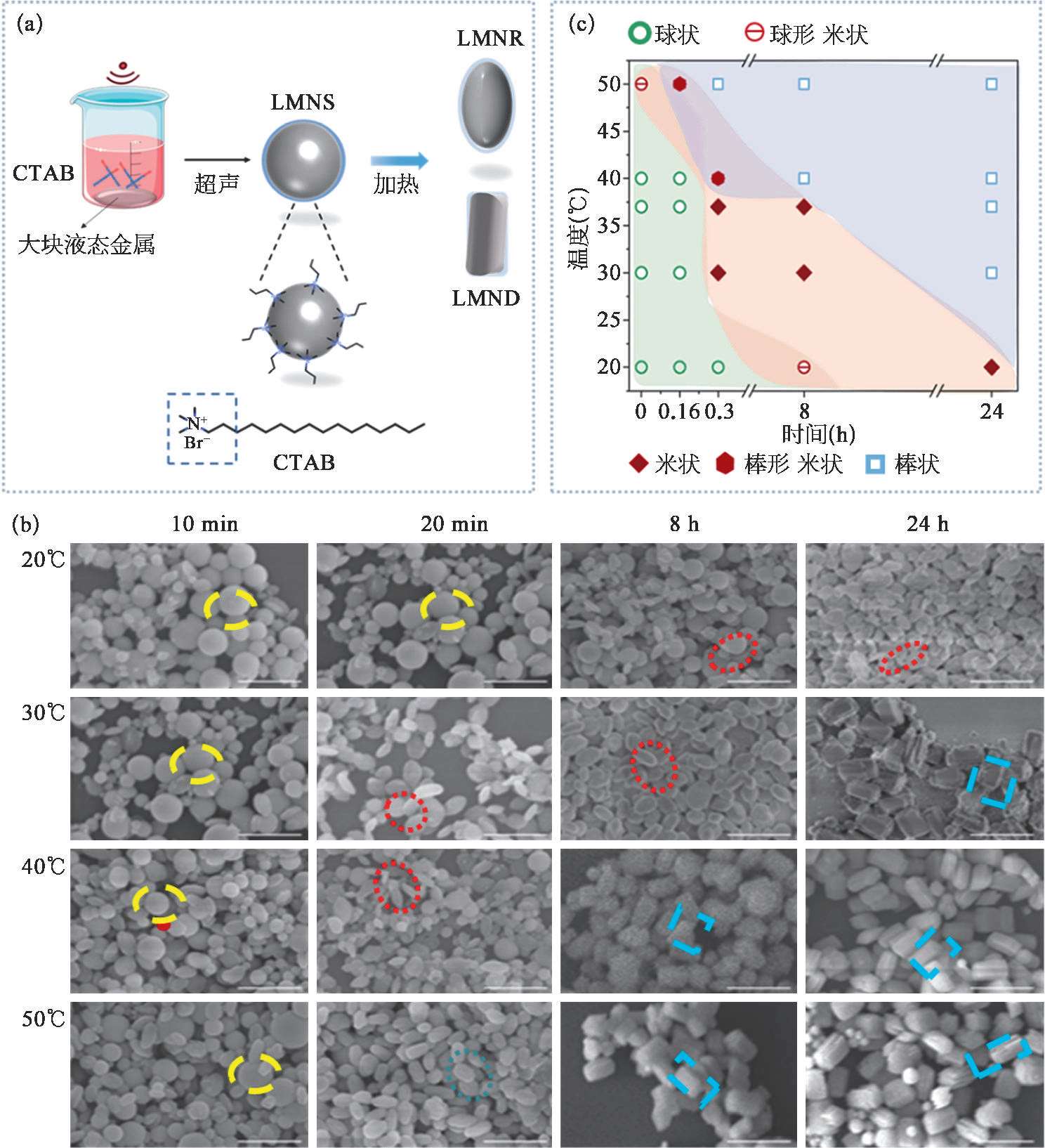
图2-10 可变形液态金属微纳米颗粒的制备与表征
(a)可变形液态金属微纳米颗粒的制备流程示意;(b)可变形液态金属微纳米颗粒在不同温度和时间下的微观形貌图(SEM);(c)球形、米状和棒状液态金属微纳米颗粒对应的时间与温度图。
在制备不同形貌的可变形的液态金属微纳米颗粒时,研究者发现烧杯中有大量气泡附着于壁面上。由文献可知 [38] ,镓基液态金属与水反应可生成棒状的GaOOH和H 2 ,其化学方程式为:
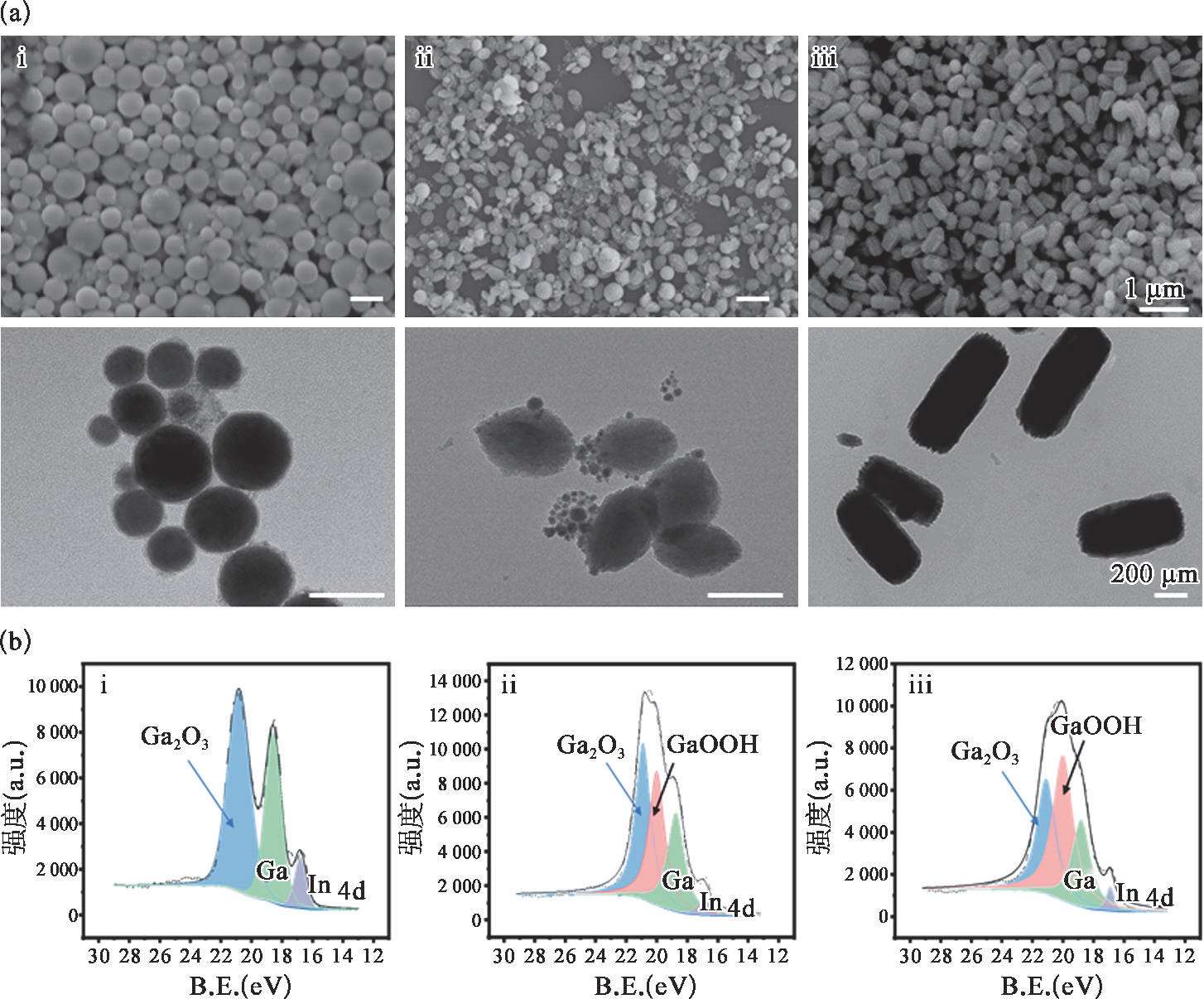
图2-11 不同形貌的液态金属微纳米颗粒的表征
(a)扫描电镜和透射电镜下的形貌图;(b)X射线光电子能谱分析图,其中(i)球形、(ii)米状和(iii)棒状。
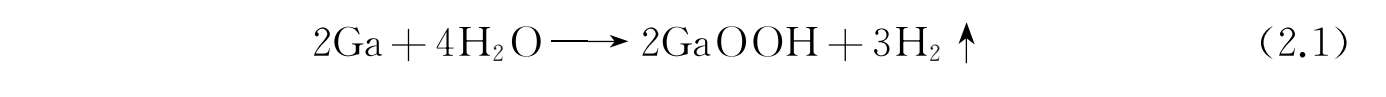
碳基材料如还原石墨烯(RGO)具有良好的导电性和离子透过性 [43] ,并且容易通过还原剂(一水合肼 [44,45] 、硼氢化钠 [46,47] 、碘化氢 [48] 、维生素C [49] 和数种金属 [50-52] )对氧化石墨烯(graphene oxide,GO)进行还原的方法制备。为了增加液态金属纳米颗粒表面的离子透过性,RGO可以作为惰性表面活性剂的合适替代品。这里以RGO包裹液态金属为例,详细讲解石墨烯液态金属纳米颗粒的制备与性能。
1. RGO包裹液态金属纳米颗粒的制备方法
将Ga、In按照EGaIn的比例(Ga,75.5%;In,24.5%)在80℃下加热搅拌2 h。将GO加入去离子水中通过预超声30 min,配制浓度1.0 mg/mL的GO溶液。然后将0.6mL的EGaIn和20mL浓度为1.0 mol/L的HCl加入盛有GO溶液的烧杯中。利用超声破碎探头对混合溶液分别进行超声时长10~30 min的超声破碎,制备得到RGO包裹的液态金属纳米颗粒(LM@RGO)。LM@RGO的制备流程展示如图2-12。
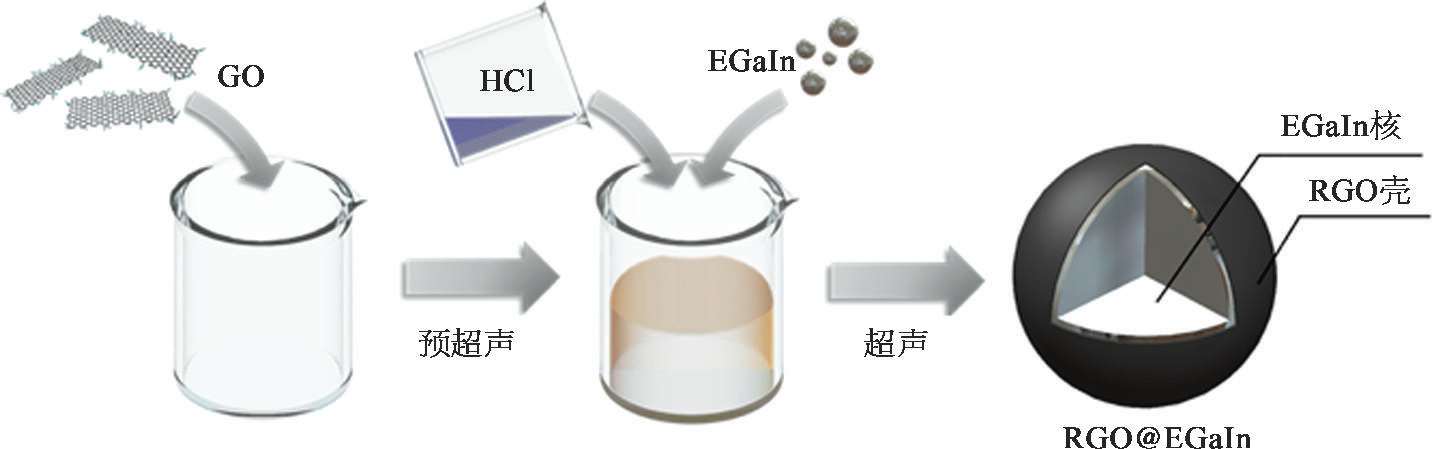
图2-12 RGO包裹的液态金属纳米颗粒的制备流程 [53]
2. LM@RGO的材料表征方法
将超声制备的液态金属纳米颗粒转移至去离子水中,滴在干净的硅片表面,并在30℃下真空干燥。在扫描电子显微镜(HITACHI S-4800)下,对LM@RGO的形貌和尺寸进行观察和统计。另外,将含有LM@RGO的溶液滴在超薄碳网上,利用透射电镜(JEM-2100)对LM@RGO的形貌进行观察,透射电镜的操作电压为200kV。同时,利用透射电镜配备的能量色散X射线光谱(EDS)对LM@RGO的组成元素进行分析。使用拉曼光谱仪(Via- Reflex)对GO和LM@RGO的RGO外壳的拉曼光谱进行测量和记录,其激光光源的波长为532nm。使用电子光谱仪(ESCALab220i-XL)分析GO和LM@RGO的RGO外壳的官能团,对GO的还原程度进行分析。在XPS测试中,使用300W的Al Kα辐射源。采用衍射仪(Bruker D8 Focus diffractometer)对离心后的样品进行晶体结构分析。采用动态光散射系统(Zetasizer Nano ZS)对不同pH的溶液中固体Ga纳米颗粒的Zeta电势进行测量,其中溶液的pH采用pH计(PHS-3C)测量得到。
3. LM@RGO的结构和组成
为了制备纳米尺寸的LM@RGO,少量的液态金属(EGaIn)被注入预超声的氧化石墨烯HCl溶液。在pH小于3的HCl溶液中,液态金属氧化膜被溶解 [54] 。通过超声方法可以实现液态金属纳米颗粒的制备,其制备流程如图2-13所示。由于EGaIn的熔点为15.5℃,通过超声方法制备的液态金属纳米颗粒仍保持液态,而高度动态且均匀一致的液态金属纳米颗粒表面将为化学反应提供更多的活化位点。从扫描电子显微成像(SEM)中可以看出,由于液态金属具有较大的表面张力 [55] ,经超声得到的液态金属纳米颗粒为了保持最小化表面能呈现出球形形貌,如图2-13所示。
利用透射电镜(TEM)和能量色散X射线光谱(EDS)可以证明,LM@RGO呈现出明显的壳核结构,如图2-14所示。在LM@RGO的组成元素中,Ga、In分布在纳米颗粒的内部,而C和O分布在EGaIn核心的外表面,即LM@RGO由EGaIn内核和RGO外壳组成。在制备LM@RGO的过程中,GO可以在超声过程中完成表面活性剂的功能,实现液态金属纳米颗粒在酸性溶液中的有效分散。

图2-13 LM@RGO的扫描电镜图 [53]
(a)和(b)的标尺为1μm;(c)和(d)的标尺为4μm。
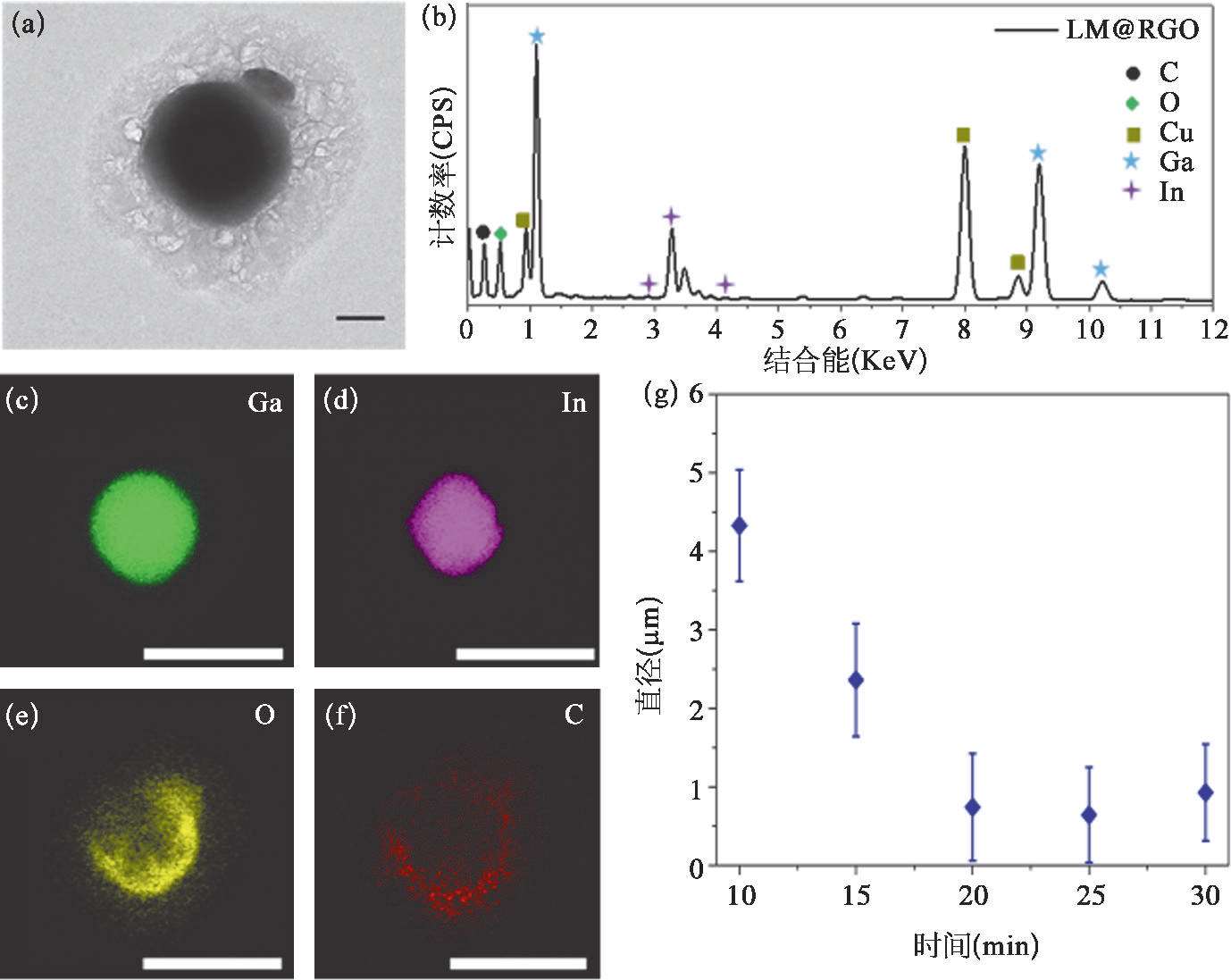
图2-14 LM@RGO的壳核结构和组成 [53]
(a)LM@RGO的透射电镜图,标尺为200nm。(b)LM@RGO的元素组成,它由Ga、In、C和O组成。Cu来自透射电镜所用的碳网。(c)~(f)LM@RGO的元素面扫描图,从左至右分别为Ga、In、O和C,标尺为600nm。(g)LM@RGO的平均粒径随超声时长的变化。
利用超声制备液态金属纳米颗粒时,超声时长将直接影响液态金属纳米颗粒的粒径大小 [56] 。当超声时长从10 min增加到20 min时,超声得到的LM@RGO的平均粒径随着超声时长的增加而减小;当超声时长超过20 min时,LM@RGO的平均粒径将基本保持稳定,如图2-14(g)所示。因此,超声20 min可以作为制备LM@RGO的标准超声时长,并使用超声20 min制备的LM@RGO进行相关性能的研究。
4. LM@RGO的性能与表征
GO的还原提高了LM@RGO外壳的电导性,有利于液态金属纳米颗粒电化学置换反应的进行。利用拉曼光谱和X射线光电子能谱(XPS),可以验证液态金属对GO的还原作用。从拉曼光谱中可以发现,如图2-15(a)所示,GO的D带与G带的峰的比值为0.86,然而LM@RGO外壳具有更高的D带与G带的峰的比值,为1.15。这主要是由于含氧官能团去除后留下的缺陷 [50] 以及小尺寸石墨化结构的生成 [57,58] 造成的。另外,GO的G带位于~1 595 cm -1 处;而在液态金属还原作用下,LM@RGO外壳的G带发生红移,偏移至~1 585 cm -1 处。这个现象说明了RGO外壳的电子态发生了改变 [59-61] ,可能与超声过程中Ga离子的插入有关。由于电子从Ga转移至RGO外壳,低浓度Ga的掺杂促使了RGO呈现出n型掺杂特性 [62] 。Ga离子在RGO层中的掺入也可以从Ga 3d轨道的XPS曲线中观察到,如图2-16所示。利用XPS的C1s和O1s的峰面积的比值同样可以反映镓基液态金属对GO的还原程度。从图2-16中可以看到,GO的C/O比值为2.17,而经过液态金属还原后的LM@RGO外壳的C/O比值可以增加到5.42。这也证明了镓基液态金属可以实现GO的部分含氧官能团的去除。另外,从C1s的XPS能谱中可以发现,相对于GO,RGO外壳的C═C键(位于284.6 eV)所占的比重明显高于GO中的C═C键的比重,RGO中的含氧官能团[位于286.6 eV的C—O键;位于287.8 eV的C=O键;位于289.0 eV的C(O)O键]的峰强度明显降低,如图2.5(b)所示。这直观地说明了GO的部分含氧官能团被镓基液态金属成功去除。从Ga 3d和In 3d的XPS曲线中可以看出,在LM@RGO的浅层存在Ga 3+ 、Ga 0 和In 0 。由于RGO外壳较厚,且XPS方法的采样深度(<10nm)较浅 [63] ,其中的Ga 3+ 信号来源于插入RGO外壳的Ga离子,而不是源自液态金属核心处。另外,如图2-15中所示,LM@RGO拉曼光谱的G峰偏移也印证了这一推测。这可以说明,镓基液态金属对GO具有一定的还原作用,且RGO包裹将对液态金属的氧化产生抑制作用。
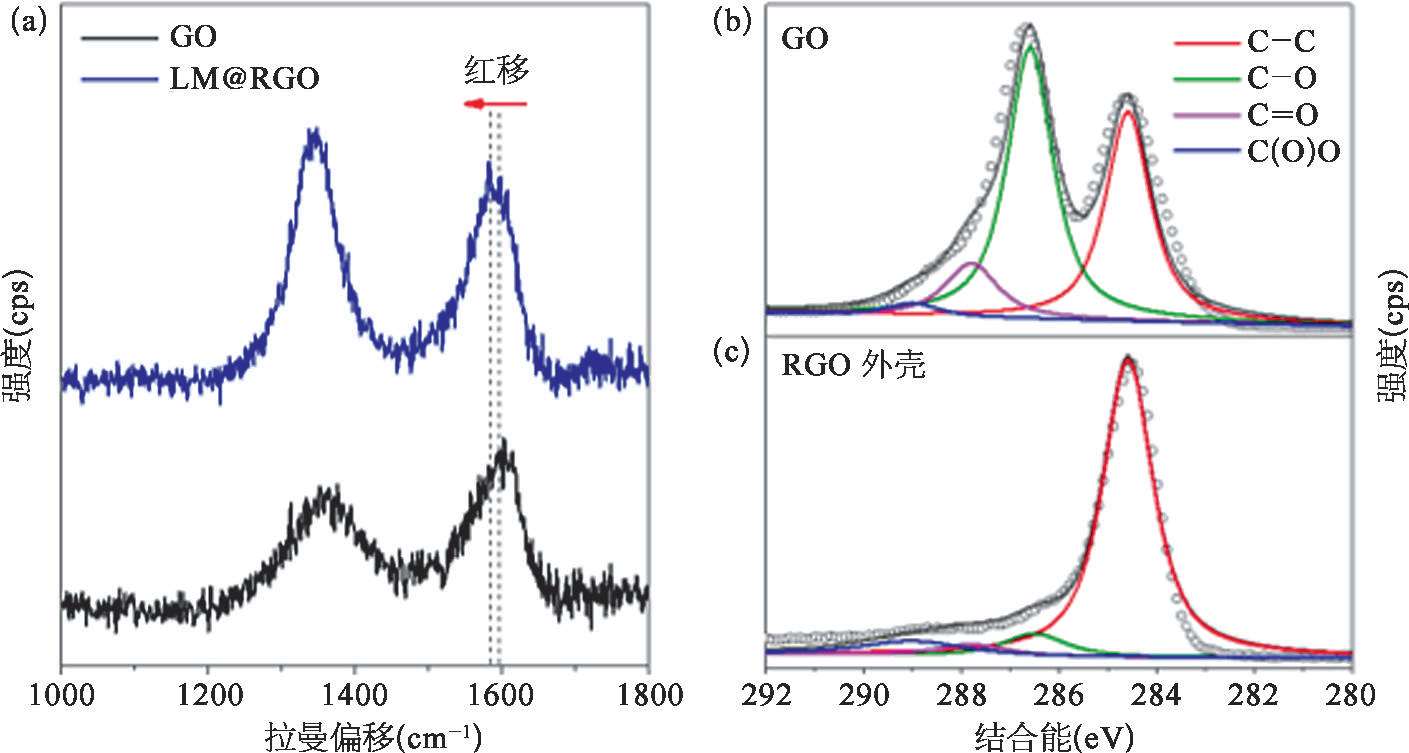
图2-15 RGO外壳的还原程度 [53]
(a)GO和RGO外壳的拉曼光谱分析结果;(b)~(c)GO和RGO外壳在C1s轨道的XPS曲线。
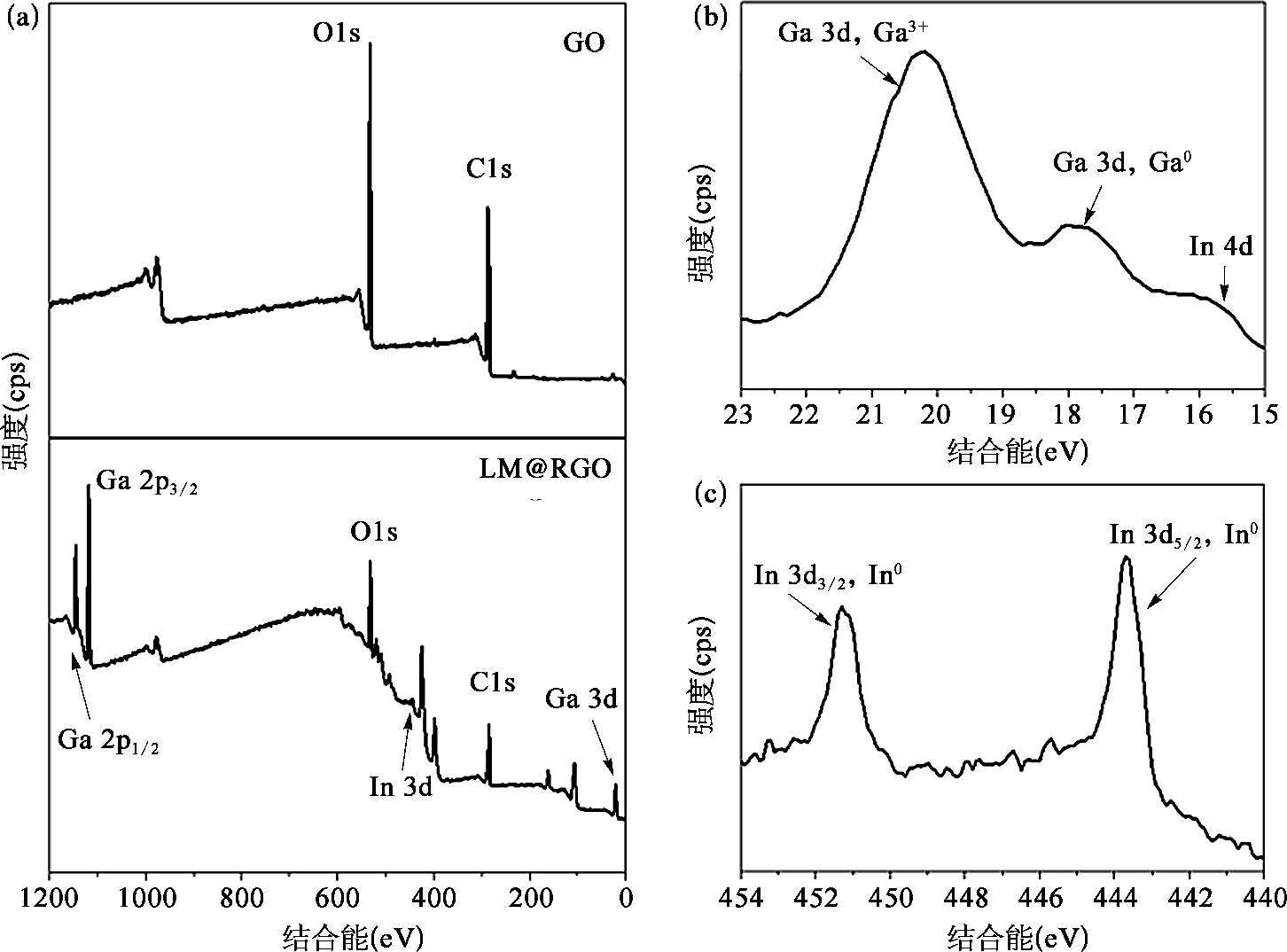
图2-16 GO和LM@RGO的XPS测量光谱 [53]
(a)对全部元素区域内的GO和LM@RGO的XPS光谱的对比;(b)LM@RGO在Ga3d轨道的高分辨率的XPS光谱;(c)LM@RGO在In3d轨道的高分辨率的XPS光谱。
镓基液态金属十分容易被氧化,即使在很低的氧气分压下也容易形成氧化膜 [64] ,并且能够被进一步氧化成GaOOH [39] 。为了证实RGO外壳可以阻止液态金属进一步氧化,将LM@RGO转移至去离子水中,并采用能谱分析LM@RGO的元素组成和分布。从TEM的能谱图中可以看出,LM@RGO由Ga、In、C和O组成,其中Ga和In主要均匀分布在液态金属核心位置,而O和C分布与Ga分布不一致,故LM@RGO表面的O主要来源于RGO而非GaO,这暗示了RGO外壳具有抑制液态金属内核进一步氧化的作用。
为了进一步揭示RGO的抑制氧化膜生成的能力,需要对RGO外壳破坏前后液态金属纳米颗粒的氧化程度进行对比分析。将LM@RGO转移至去离子水中,在转速4 000 r/min下进行快速离心,在离心力的作用下可以将LM@RGO的还原石墨烯外壳破坏。为了验证离心前后LM@RGO的变化,利用SEM和TEM对离心前后的样品形貌进行观测,并利用X射线衍射(XRD)对离心前后的样品组成进行分析。从图2-14(c)至图2-14(f)中可以看出,离心前的LM@RGO样品的主要组成元素为Ga和In。从图2-17(a)和图2-17(b)中可以看出LM@RGO经离心后形貌由球形转变成短棒状,且颗粒的平均尺寸接近或略小于液态金属纳米颗粒的尺寸,颗粒尺寸的略微减小主要是由于In的析出导致。相似的现象可以在Ga的电化学氧化过程中观察到 [65] 。在离心后,短棒状纳米颗粒的O含量明显增加,如图2-17(c)所示,这也说明了当RGO外壳破坏后,液态金属纳米颗粒在溶液中被迅速氧化成GaOOH。当RGO外壳被破坏时,由于电子可以快速从液态金属转移至RGO上,液态金属纳米颗粒极易被氧化,这已经在液态金属在石墨表面发生氧化变形的研究中被证实 [66] 。因此,当离心作用破坏RGO外壳后,液态金属纳米颗粒将发生化学转变,被氧化形成GaOOH,从而呈现出短棒状结构。由于液态金属颗粒纳米化后表面增大,这将加剧液态金属纳米颗粒的氧化,如图2-17(d)所示。在液态金属纳米颗粒中Ga被氧化成GaOOH,而In的改变并未发生。当二元EGaIn中Ga逐渐被消耗时,In从合金中被析出沉淀。从TEM图中可以发现在短棒状颗粒中存在球形颗粒,如图2-18(a)和(b)所示。经EDS分析,可以证实这些球形颗粒由In组成,如图2-18(c)和(d)所示。经过对离心后的样品进行XRD测试,可以证实离心后的样品的XRD峰分别来自GaOOH(2 θ =21.7°、33.9°、35.4°、37.4°、40.8°、54.4°和60.2°)和单质In,如图2-17(c)所示。因此,完整的RGO外壳可以对液态金属的氧化产生抑制作用,从而保护液态金属内核不被进一步氧化,这主要源于RGO对气体的低渗透性 [67,68] 。另外,由于金属离子的尺寸小于石墨烯的层间距,金属离子可以穿过石墨烯层,从而不影响金属离子的质量传输 [69] 。因此,LM@RGO可以作为电化学置换反应的合适的反应平台。
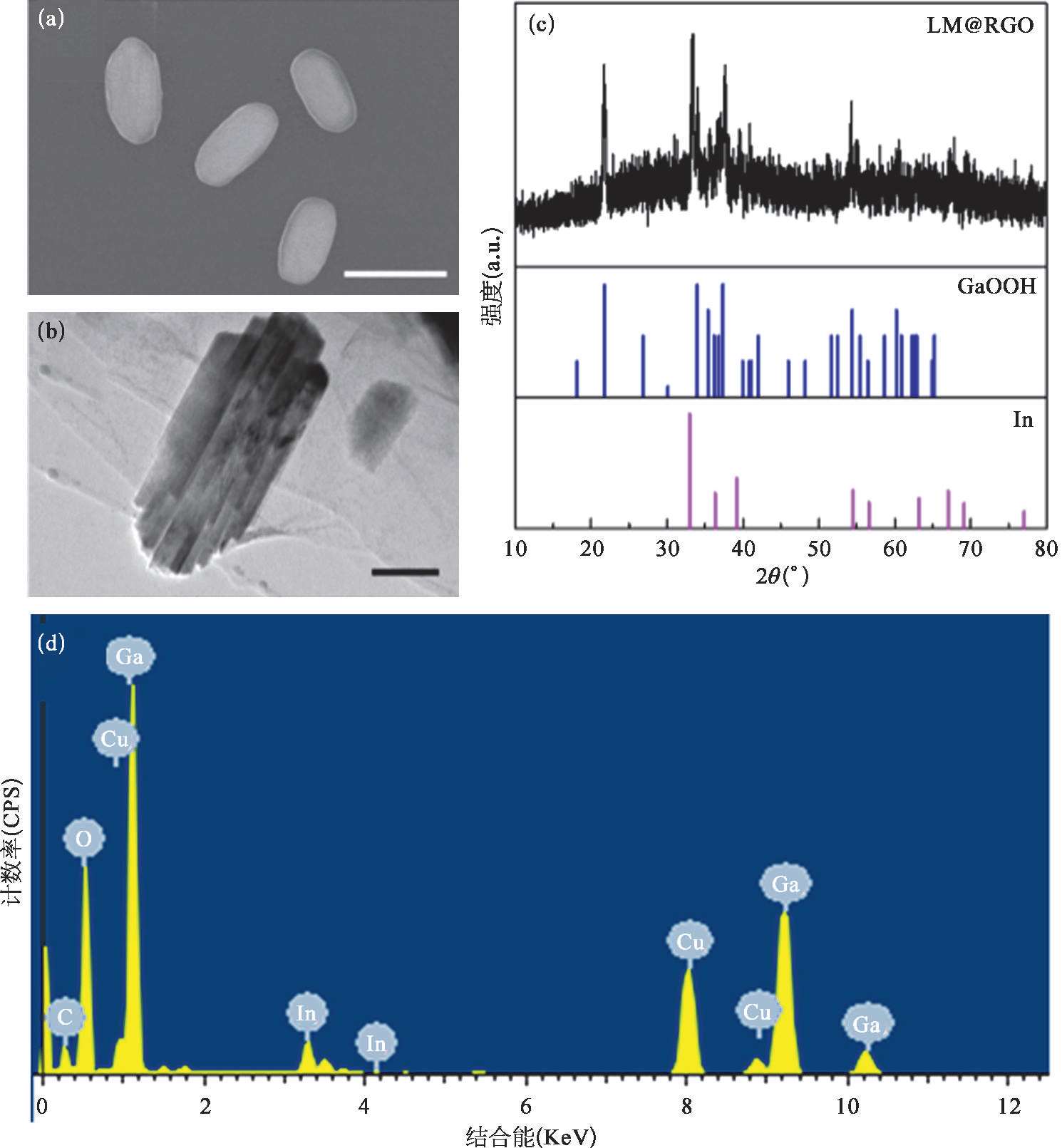
图2-17 经过高速离心后的纳米颗粒样品的形貌和氧化程度分析 [53]
(a)4000r/min高速离心后纳米颗粒的形貌电镜图,标尺为500nm。(b)离心后的纳米颗粒样品的透射电镜图。标尺为100nm。(c)离心后的纳米颗粒样品的XRD曲线以及GaOOH和In的XRD标准峰。(d)经过高速离心后的纳米颗粒样品元素组成。
5. GO与液态金属纳米颗粒的相互作用机理
在已有的研究中,液态金属的氧化膜在形成液态金属纳米颗粒的过程中具有很重要的作用。然而,由于在酸性溶液中(pH<3)氧化膜被溶解,LM@RGO的形成机理将不同于之前的研究。在LM@RGO的超声形成过程中,液态金属与GO之间的静电相互作用将起到主导作用,同时完成GO的还原。在酸性溶液中,镓基液态金属与溶液中的H + 反应,生成Ga 3+ ;由于生成的Ga 3+ 吸附在液态金属纳米颗粒表面,使得液态金属纳米颗粒带有正电 [50] 。因为GO具有丰富的官能团,使得GO带有负电 [70] 。另外,随着溶液pH的减小,液态金属纳米颗粒表面带有的正电荷增加,如图2-19所示。由于液态金属纳米颗粒与GO具有不同的电性,依靠静电相互作用可以实现将GO包覆在液态金属纳米颗粒表面,原理如图2-20所示。在图2-20的红色虚线框中,展示了GO被镓基液态金属还原的原理,还原过程中需要H + 的参与,氧化还原反应的反应式如下所示 [52] :
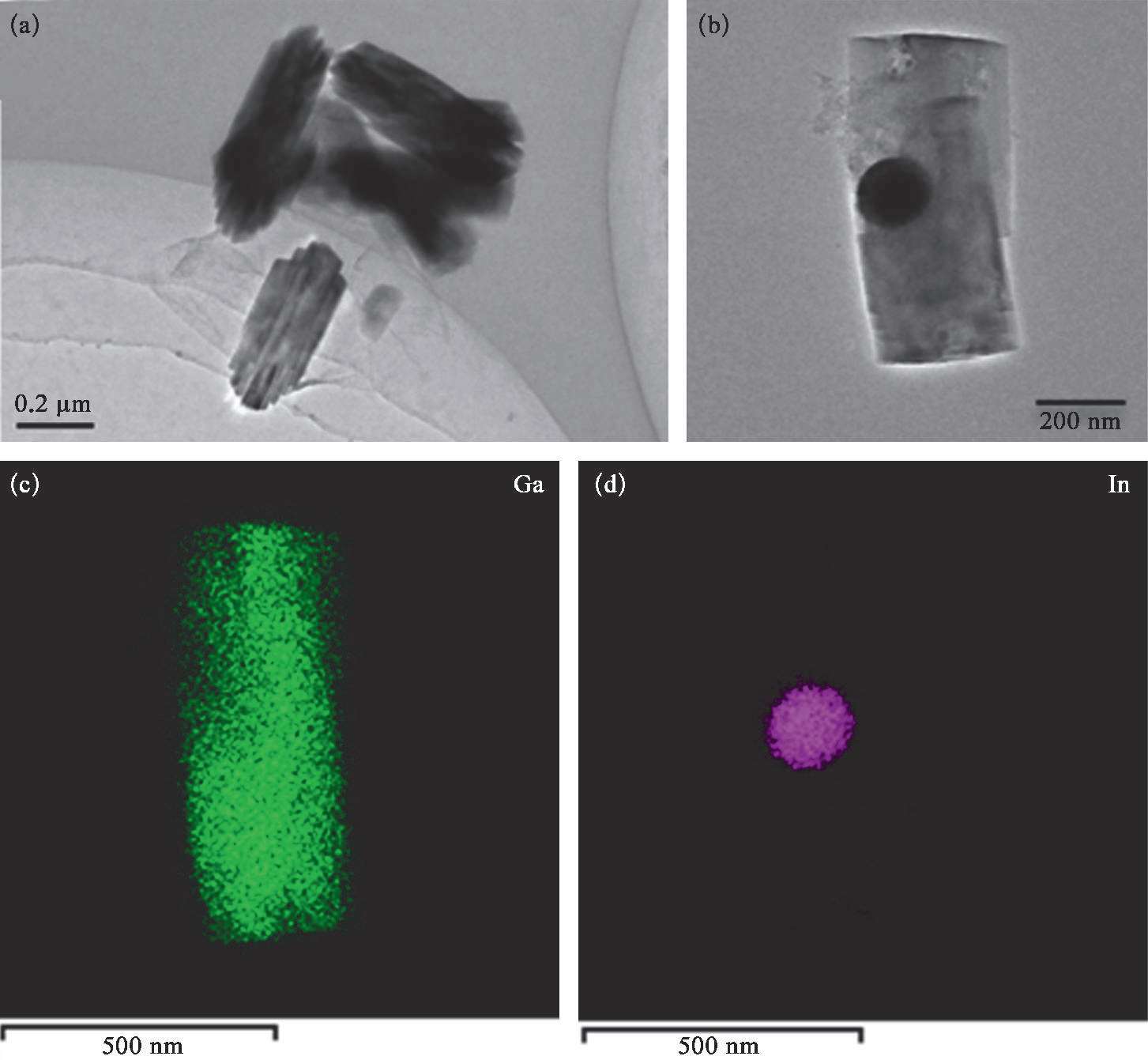
图2-18 离心后的纳米颗粒形貌和元素组成 [53]
(a)离心后的纳米颗粒样品的透射电镜图;(b)样品的放大图;(c)~(d)离心后纳米颗粒样品中Ga和In的分布。
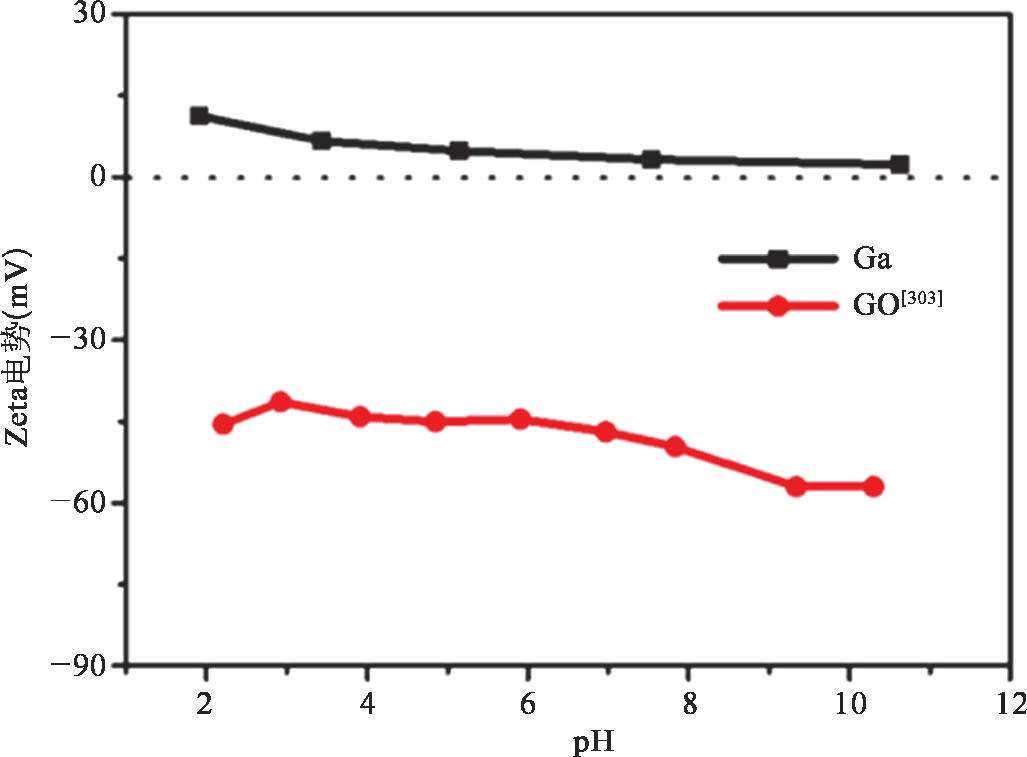
图2-19 不同pH的水溶液中GO和Ga纳米颗粒的Zeta电势变化 [53]

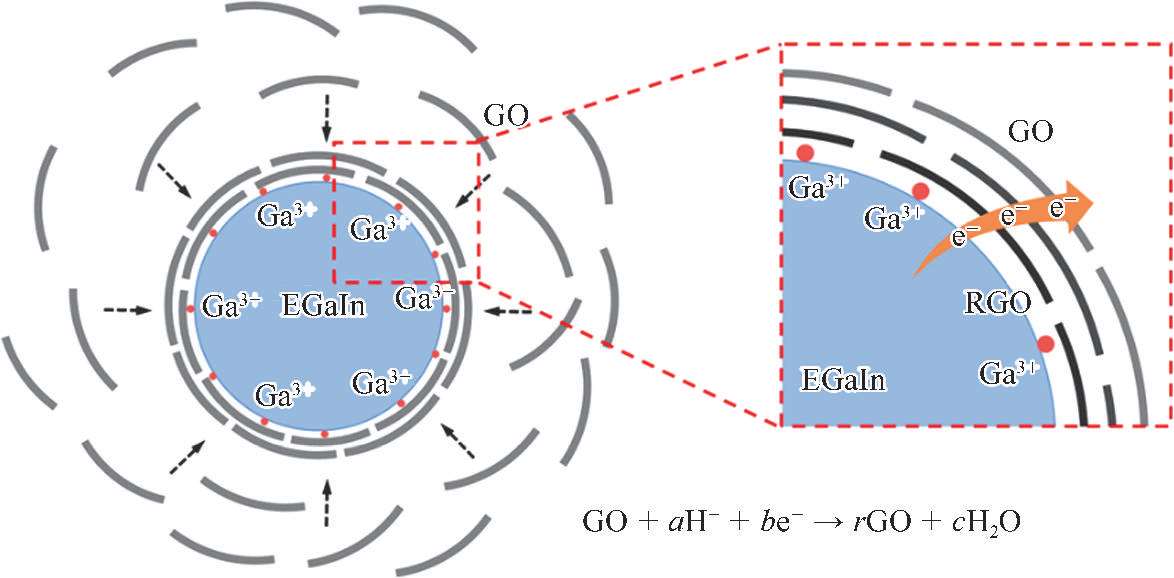
图2-20 在酸性溶液中,液态金属纳米颗粒表面发生的GO部分还原和RGO自组装的机理示意 [53]
在氧化还原反应中,由于Ga具有较强的还原性 [71] ,液态金属提供了反应所需的电子。这也可以通过对酸性溶液中Ga和GO的循环伏安曲线的分析中得到。在GO的循环伏安曲线中,如图2-21所示,可以得到,在1.0 mol/L的HCl溶液中,GO的还原电位为-1.06V,并且从-0.6V开始,GO逐渐被还原。对于Ga,在1.0 mol/L的HCl溶液中,其循环伏安曲线出现三处氧化峰,分别依次对应Ga的3种不同价态,即Ga + 、Ga 2+ 和Ga 3+ 。循环伏安曲线的主要的氧化峰出现在0.82V,代表Ga 3+ 是Ga的稳定离子状态。因此,在Ga的氧化过程中释放出的电子可以传递至GO,进而发生式(2.2)中的氧化还原反应。也就是说,Ga在酸性条件下可以部分还原GO。另外,在电子传递过程中产生的局部电场将促进GO在液态金属纳米颗粒表面的自组装 [57] 。
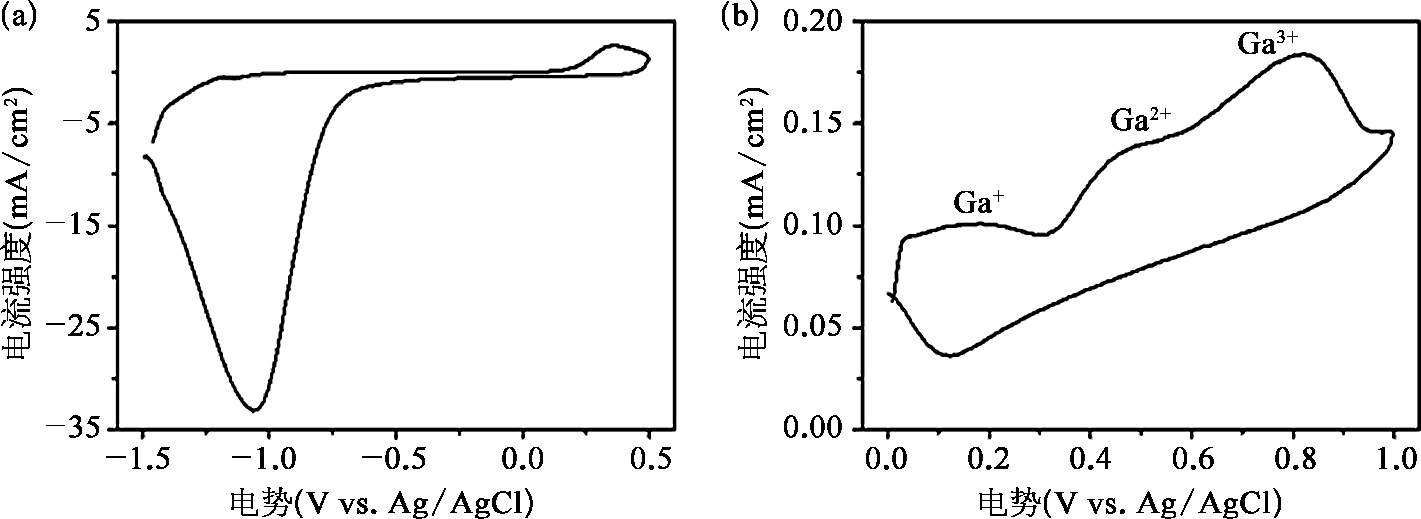
图2-21 循环伏安曲线测试 [53]
(a)在浓度为1.0mol/L的HCl溶液中GO的循环伏安曲线;(b)在浓度为1.0mol/L的HCl溶液中Ga的循环伏安曲线。
常用的无机氧化物修饰主要有SiO 2 与ZrO 2 等,本节以ZrO 2 修饰为例 [72,73] 。
1. ZrO 2 修饰纳米液态金属颗粒的制备方法
(1)LM-ZrO 2 纳米胶囊的制备
首先,将0.015mL Span 85加入60mL乙醇中并均匀混合,向混合物中添加0.105mL液态金属,通过超声波将液态金属从大液滴分散成小颗粒。然后,将20mL乙腈和1.2mL氨添加到先前的溶液中。向15mL乙醇和5mL乙腈的混合物中添加0.5mL正丙醇锆。将上述两种溶液混合在一起,并在室温下用磁力搅拌器搅拌6h。将正丙醇锆水解成ZrO 2 并与液态金属复合。通过离心收集LM-ZrO 2 纳米颗粒。
(2)IL-LM-ZrO 2 纳米晶的制备
所获得的材料有望在生物体内具有促进微波加热的特性。因此,在获得的LM-ZrO 2 纳米颗粒中负载IL,因为封闭空间中的IL具有良好的微波灵敏度。将90 mg LM-ZrO 2 NCs添加到15mL乙醇中。然后将1mL IL和2mL 1,4-二恶烷的混合物添加到先前的溶液中。用磁力搅拌器搅拌混合物约4h。通过离心收集IL-LM-ZrO 2 NCs。
(3)PEG-IL-LM-ZrO 2 纳米晶的制备
尽管液态金属毒性较低,但应考虑合成材料的毒性。由于聚乙二醇(PEG)具有良好的生物相容性,因此将其涂覆在所获得的IL-LM-ZrO 2 纳米颗粒上用于细胞实验和动物实验。将IL-LM-ZrO 2 纳米颗粒分散在10mL Tris-HCl缓冲液(pH=8)中,添加PEG-SH粉末(5kDa),并在室温下将混合物磁搅拌4h。通过离心收集PEG-IL-LM-ZrO 2 NCs。
2. PEG-IL-LM-ZrO 2 NCs的合成与表征
如图2-22中SEM和TEM图像所示,生成的LM-ZrO 2 NCs基本上是直径为(260±60)nm的球形固体纳米颗粒[图2-22(a)和(b)]。LM-ZrO 2 NCs的EDS结果表明NCs中含有O、Zr、Ga和In元素[图2-22(c)]。如图中所示,O、Zr、Ga和In元素均匀混合在一起[图2-22(d)]。结果表明,所制备的液态金属纳米材料具有粒径分布均匀、分散性好的特点。
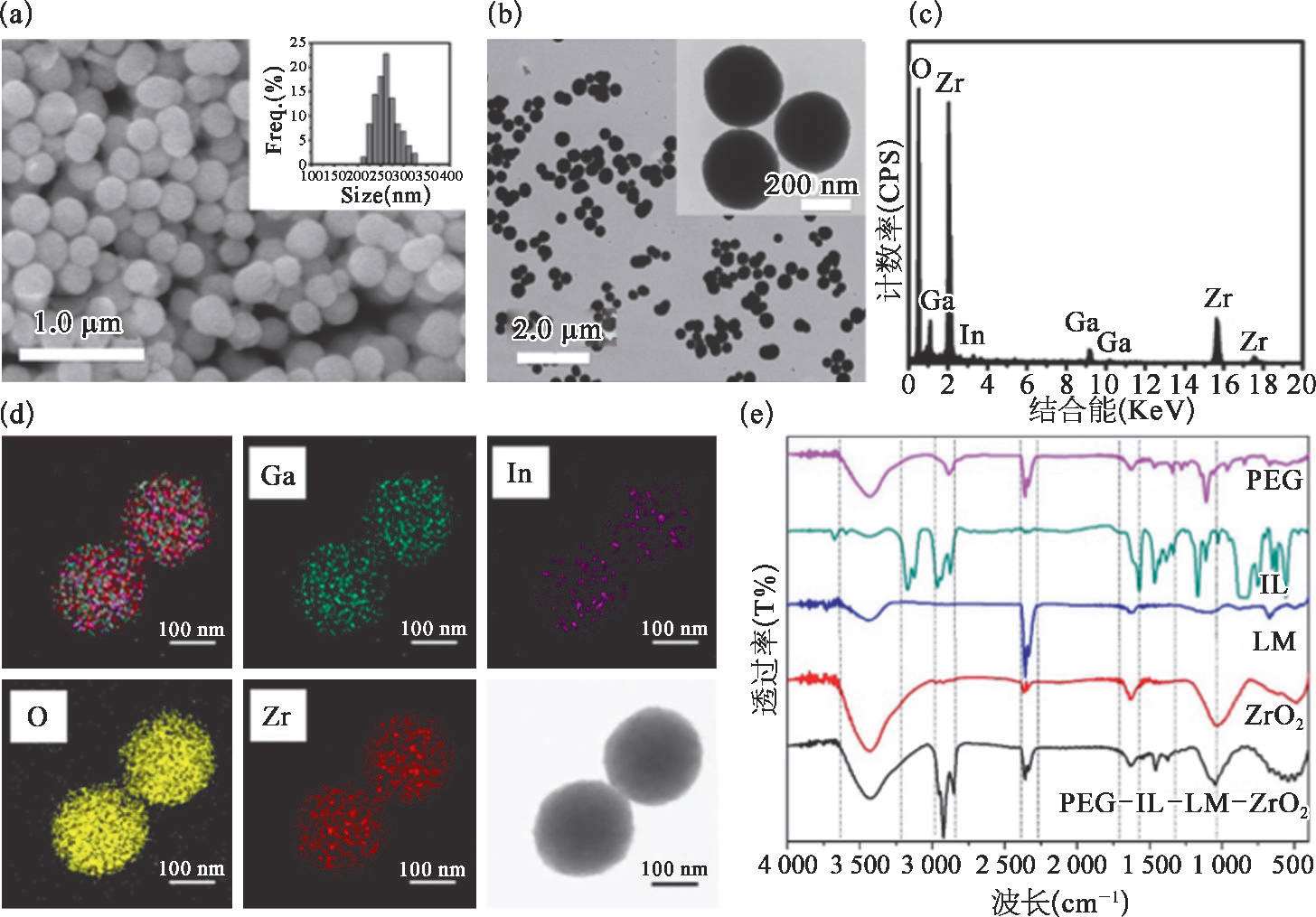
图2-22 PEG-IL-LM-ZrO 2 NCs的表征 [73]
(a)LM ZrO 2 NCs的SEM图像和粒度统计;(b)LM ZrO 2 纳米晶的TEM图像;(c)LM ZrO 2 NCs的EDS痕迹;(d)LM ZrO 2 NCs的TEM元素映射;(e)PEG、IL、LM、ZrO 2 和PEG IL LM ZrO 2 NCs的FT IR光谱。
如PEG-IL-LM-ZrO 2 NCs的FT-IR图像所示[图2-22(e)],3 550和3 200 cm -1 之间的强而宽的吸收带是O—H伸缩振动。IL中—CH 3 的不对称拉伸振动发生在2 960和1 380 cm -1 处。—CH 2 的不对称拉伸、对称拉伸和剪切振动分别发生在2 925、2 850和1 462 cm -1 处。1 050 cm -1 为C—O特征吸收的拉伸振动。这些数据表明成功合成了PEG-IL-LM-ZrO 2 NCs。