
下载掌阅APP,畅读海量书库
立即打开



来自下丘脑、垂体以及甲状腺自身反馈的信息共同调节甲状腺激素的合成,垂体分泌的促甲状腺激素(Thyroid Stimulating Hormone, TSH)是甲状腺生长和功能的主要调节因素。TSH是由位于垂体前叶的TSH分泌细胞合成的糖蛋白激素,由α亚单位和β亚单位组成。α亚单位由6号染色体上的α基因编码,为TSH、FSH、LH、CG等糖蛋白激素共有;β亚单位由1号染色体上的TSHβ基因编码,决定TSH分子的结构特异性。TSH主要与甲状腺滤泡上皮细胞上的促甲状腺激素受体(TSHR)结合而发挥其促进甲状腺激素合成的作用。基础分泌模式和脉冲分泌模式的协调形成了TSH分泌的昼夜节律。TSH合成和分泌的过程受到中枢与外周因素的精密调控,下丘脑合成的促甲状腺激素释放激素是促进TSH合成、分泌的主要因素且可能参与TSH调定点的控制;T 3 、T 4 可负反馈抑制下丘脑、垂体的活动,是抑制TSH合成、分泌的主要因素。敏感且特异的TSH测定技术对甲状腺疾病的诊断和指导治疗意义重大,免疫计量分析法是目前广泛应用的TSH测定方法。年龄、碘营养水平等因素会对TSH水平产生影响,人群特异性的TSH参考范围有待探讨及制定。人重组TSH分子已成功合成并获得一定的临床价值。
本节将从垂体器官的发育,TSH细胞的发育、解剖和细胞学,TSH分子结构及亚单位编码基因,TSH合成,TSH分泌,TSH合成调节,TSH分泌调节,TSH的作用,TSH测定,年龄与TSH,重组人TSH等方面进行阐述。
垂体由垂体前叶、后叶及退化的中叶组成,位于蝶鞍内,垂体柄穿过硬脑膜与下丘脑正中隆起相连。垂体起源于口腔神经板,人类胚胎3周左右,口腔前庭的原始外胚层凹陷形成Rathke's囊,是腺垂体的起源,腺垂体的细胞分化主要在前12周完成。Rathke's囊直接连接到垂体柄和下丘脑漏斗部,最终从口腔和鼻咽分离出来,突向第三脑室并与脑室憩室融合,随后空腔消失。Rathke's囊形成腺垂体,神经垂体随着第三脑室发育从神经外胚层发育而来。
腺垂体的细胞可根据其分泌激素的种类分为不同的细胞群,促肾上腺皮质激素细胞分泌包括促肾上腺皮质激素(Adrenocorticotrophic Hormone,ACTH)在内的阿片-促黑素细胞皮质素原(Proopiomelanocortin, POMC)肽类,生长激素细胞分泌生长激素(Growth Hormone, GH),促甲状腺激素细胞(Thyrotrophs)分泌糖蛋白激素α亚单位和促甲状腺激素特有的TSHβ亚单位,促性腺细胞分泌卵泡刺激素(Follicule-Stimulating Hormone,FSH)和黄体生成素(Luteinizing Hormone, LH),催乳素细胞分泌泌乳素(Prolactin, PRL)。胚胎发育过程中,这些不同的激素分泌细胞都起源于Rathke's囊前体细胞,垂体的发育过程是前体细胞终末分化为不同细胞类型的经典发育模式 [1] 。
1.垂体器官形成的信号和转录机制:脊椎动物从胚胎发育、器官发生到形成完整生命体的过程受到复杂的信号通路的精密调控,下丘脑-垂体轴被认为是连接大脑与外周激素分泌器官的神经内分泌传感器,其发育过程中也需要一系列转录因子及信号通路的参与。垂体的形态发生需要很多重要的神经外胚层信号参与,包括初始囊内陷所需要的漏斗骨形态生成蛋白4(BMP4)、成纤维细胞生长因子8(FGF8)、Wnt5和Wnt4;接下来腹侧的形态发展及转录因子的表达则由BMP2以及音猬蛋白(Shh)进行调控,它们的空间和梯度表达对决定细胞增殖的早期模式至关重要。
(1)骨形态生成蛋白(Bone Morphogenetic Protein, BMP):BMP是调控早期垂体发育不可或缺的调节因子,BMP4和BMP2参与垂体前叶的发育。BMP4属于转化生长因子β(Transforming Growth Factor, TGFβ)超家族,与口腔外胚层分化为垂体过程中的细胞分化塑形密切相关。胚胎发育第8.5~9天,BMP4蛋白在腹侧间脑表达,继而在漏斗区表达,垂体发育后期表达水平降低。口腔外胚层和腹侧神经外胚层内陷形成Rathke's囊的起始步骤依赖于BMP4的存在,BMP4阻断将导致垂体发育停滞以及垂体中所有激素分泌细胞的缺失。BMP2控制垂体腹侧多种转录因子的表达,也控制糖蛋白激素α亚单位基因的表达,α亚单位基因表达是TSH细胞和促性腺细胞分化早期的标志性信号。BMP2信号途径使祖细胞向特异的激素分泌细胞分化,并调节各种细胞类型在垂体腹侧的区域定位 [2] 。
(2)音猬蛋白(Sonic Hedgehog, Shh):分泌因子Shh在器官整体发生过程中起到重要作用,Shh敲除导致胚胎轴向发育异常、Rathke's囊缺失,然而这种现象可能源于间脑/漏斗中线结构的缺失而不是前叶垂体原基的缺失。小鼠垂体发育过程中,Shh在间脑腹侧和口腔外胚层表达,而在Rathke's囊形成的区域并无表达。Shh可能在BMP2信号通路级联反应中发挥作用,决定垂体腹侧的细胞系谱。锌指转录因子Gli1、Gli2、Gli3可能是Shh信号通路的下游分子,灭活Gli1/Gli2会导致发育过程中垂体缺失。
(3)成纤维细胞生长因子(Fibroblast Growth Factor, FGF):器官形成过程中,FGF家族的不同成员在细胞类型分化的不同阶段发挥不同的生物学作用。垂体发育早期,FGF8、FGF10、FGF18在间脑腹侧和垂体后叶表达,FGF8和FGF10控制垂体细胞增殖和不同细胞类型的区域限制性分布。FGF10受体(FFGRⅡb)条件性敲除的小鼠模型,Rathke's囊初步形成后很快发生凋亡,导致胚胎第14.5天时垂体器官发育不全;这提示FGF10信号通路调节发育过程中的细胞存活/凋亡 [3] 。FGF8阻断导致发育第8.5天时胚胎死亡,先于垂体器官形成。漏斗管形成可刺激细胞增殖,促进Lbx3表达,FGF8可部分模仿漏斗管的生物活性模式,FGF8过表达可使Lbx3表达增强和垂体增生。总的来说,FGF8的主要功能是维持细胞增殖,对抗BMP2的分化信号。
(4)Notch信号途径:目前已知的调节垂体原基分化起始过程的信号通路是Notch通路。Notch基因编码一类高度保守的细胞表面受体,由Notch受体、Notch配体(DSL蛋白)及细胞内效应器分子(CSL-DNA结合蛋白)3部分组成,调节从海胆到人类等多种生物的胚胎发育。目前研究较为明确的Notch途径靶分子是HES家族成员,包括HES1、HES5。垂体发育过程中,Notch途径与HES1、HES5参与垂体祖细胞的维持和垂体中叶以及后叶的发育调控。Notch途径直接调节转录因子PROP1的表达,PROP1的出现是表达Pit-1的前体细胞形成的必要条件。随后,Notch信号在Pit-1前体细胞中的衰减对不同谱系的Pit-1垂体细胞的形成是必要的,促甲状腺激素分泌细胞和促性腺激素分泌细胞内过表达Notch将导致细胞发育缺陷,异常Notch激活导致的发育缺陷与bHLH转录因子Mash1、Mash3的表达降低有关 [4] 。Mash1是促甲状腺素分泌细胞、促性腺激素分泌细胞、促糖皮质激素分泌细胞和促黑素分泌细胞终末分化的执行者,垂体中Mash3的表达需要Pit-1的调节,从胚胎发育第13.5天持续到成年,负责调节对GHRH的反应。
(5)Wnt/β-连环蛋白信号途径:Wnt蛋白家族是一种分泌性的信号分子,在发育过程中参与胚胎诱导、细胞命运决定、稳态维持。经典Wnt/β-连环蛋白通路的激活使β-连环蛋白稳定性增加,继而向核内转位作为DNA转录因子结合域的共活化因子发挥生物学作用;多数情况下,Lef/T通过置换HDAC、TLE/groucho共阻遏物和募集p300/CBP、Brg1,促进靶基因表达。Wnt/β-连环蛋白还参与Pit-1决定的前体细胞谱系分化和垂体的生长,因此Wnt/β-连环蛋白信号通路从胚胎第11.5~15.5天在垂体中是处于激活状态的,可通过检测其下游直接的靶基因Axin2评估其活性。短暂的Wnt/β-连环蛋白控制对正常的垂体发育是必要的,胚胎第13.5天时,β-连环蛋白不充分激活导致的Hex 1抑制可使垂体发育不全。Wnt/β-连环蛋白信号通路的其他成员,如Fzd2、Lef1、Tcf3、Tcf4,在垂体发育过程中也有表达。胚胎第12.5天时,在Rathke's囊和漏斗中可检测到Fzd2;胚胎第9~14.5天可在垂体中检测到Tcf3,而且只局限于有Pit-1表达的体部;Tcf4在发育早期的垂体及周围组织中可检测到,胚胎第13.5天以后显著降低;Lef1有两个表达时相,胚胎第9天时在Rathke's囊短期表达,胚胎第13.5天时在垂体前叶和中叶再次出现。灭活Tcf4导致垂体前叶增生,灭活Lef1导致Pit-1、GH、TSHβ表达增加 [5] 。Wnt家族的其他成员在发育中的垂体也可检测到,其中Wnt4、Wnt5与垂体前叶的发育密切相关。
2.控制垂体发育早期阶段的转录因子:各种类型的腺垂体细胞的功能发展涉及多能垂体干细胞表达的细胞系特定转录因子动态梯度的时空调节,生长激素细胞、促甲状腺激素细胞、催乳素细胞都来源于表达转录因子Pit-1的前体细胞,Pit-1是垂体特有的转录因子,其表达依赖于与其成对出现的转录因子PROP1的存在;而促肾上腺皮质激素细胞的终末分化依赖于转录因子Tbx19(T-box Transcription Factor)。因此,全能干细胞分化为腺垂体细胞系的主要决定因子是PROP1,因为它决定了后续Pit-1依赖性细胞系的发育和促性腺激素细胞系的发育。
垂体早期细胞分化需要细胞外Rpx和Ptx的表达。如图4-1所示,Rathke's囊表达LIM同源异型结构域激素家族的多个转录因子,包括Lhx3、Lhx4、ISL1,它们对垂体功能发育早期具有重要作用。Ptx是广泛的垂体调节因子,也能活化α糖蛋白亚单位(αGSU)、LHβ(Ptx1)及GH(Ptx2)的转录。Lhx3能决定GH、PRL和TSH细胞的分化。PROP1是Pit-1的先决条件,能够活化GH、PRL、TSH和促生长激素释放激素受体(GHRHR)的转录。TSH细胞和促性腺激素细胞表达共同的α糖蛋白亚单位(αGSU),其表达受Gata2调控。这些转录因子时间与空间的表达变化参与调控形态和功能各异的激素分泌细胞的形成。
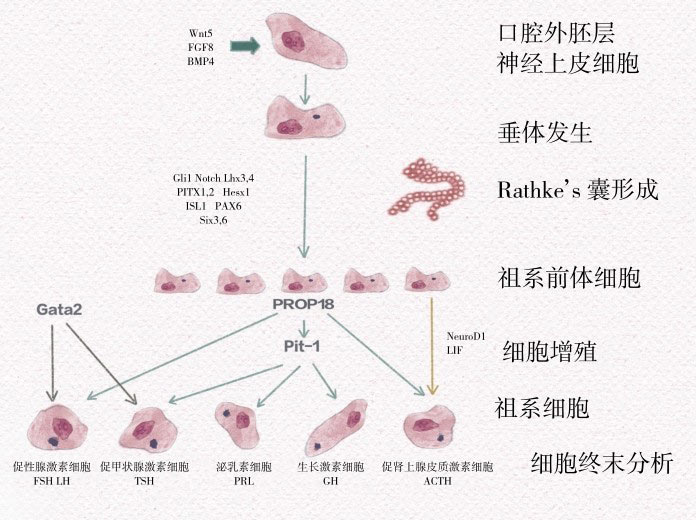
图4-1 口腔外胚层神经上皮细胞增殖、分化形成Rathke's囊的过程需要Lhx3、Lhx4、ISL1等多种转录因子的精密调控,对垂体祖系前体细胞形成及垂体功能早期发育具有重要作用。PROP1是垂体特有转录因子Pit-1的主要调控因子,它决定了由垂体祖系前体细胞到腺垂体各功能性细胞系的分化和发育过程。TSH细胞和促性腺激素细胞发育过程还受到DNA结合元件Gata2的调节
(1)PITX:目前已有3个PITX相关的转录因子被发现,它们与Bicoid(以前又叫作P-Otx或PITX1)共有同源结构域。PITX3对哺乳动物中脑多巴胺能神经元的正常发育至关重要。PITX1和PITX2基因在几乎所有类型的垂体细胞中表达,并持续整个发育过程;PITX1和PITX2的表达分布区域虽有重叠但并不相同,这两个转录因子呈剂量依赖性促进细胞增殖和分化,PITX2的生物学功能相对更强大,PITX1或PITX2都能控制Lbx3的表达。PITX1于垂体器官形成早期在神经板前区和口腔外胚层表达,可与垂体特有的转录因子Pit-1的氨基末端结构域相互作用。目的性破坏PITX1结构导致垂体促甲状腺素分泌细胞和促性腺激素分泌细胞的终末分化标志减低 [6] 。PITX2基因是在人Rieger综合征的基因定位克隆过程中发现的。PITX2基因敲除小鼠有很多先天性缺陷,包括Rathke's囊和漏斗部连接建立后的早期垂体发育停滞,垂体发育过程各种信号分子梯度的形成也出现异常。PITX相关的转录因子与αGSU、TSHβ、LHβ、FSHβ、GH、PRL、GnRHR基因启动子结合调节其转录过程。
(2)LIM同源域蛋白(The LIM-Homeodomain Proteins, LIM-HD):LIM-HD蛋白分子在N-末端与DNA结合同源域间含有两个串联衔接的LIM结构域,LIM结构域为LIM-HD分子与其所募集的辅酶因子提供蛋白-蛋白相互作用的位点,从而完成LIM-HD生物学活性的发挥。Rathke's囊表达LIM-HD家族的多个转录因子,包括Lhx3、Lhx4、ISL1,它们对垂体功能发育早期具有决定性作用。胚胎发育第9.5天时,Lhx3在Rathke's囊特异表达,它与Lhx4协同作用在垂体早期器官形成过程中发挥决定性作用。Lhx3基因敲除鼠Rathke's囊退化,外胚层细胞增殖障碍,除促肾上腺皮质激素分泌细胞外,其他垂体细胞类型全部缺失。漏斗FGF8诱导的Lhx3的表达在口腔外胚层分化为垂体的细胞选择过程中是关键的步骤,Lhx3可与PITX2协同作用激活垂体特异基因α-GSU的表达。胚胎发育第8.5天,ISL1最初在口腔外胚层表达;胚胎第9.5天在Rathke's囊表达;胚胎第10.5天,ISL1的表达局限于Rathke's囊的腹侧,与喙部聚集的促甲状腺激素细胞共定位。BMP4、BMP2是ISL1基因表达的激活因素,而FGF8/FGF2抑制其表达,两种信号分子分别在垂体背、腹侧对ISL1的表达进行反相调节,由此形成空间上的浓度梯度对垂体发育意义重大。ISL1基因缺陷鼠胚胎第10天时,出现心脏、胰腺、运动神经元等多处发育缺陷,原始Rathke's囊形成但上皮细胞发育畸形,这提示ISL1对于垂体祖细胞的增殖和分化至关重要。
(3)Hesx1:转录因子Hesx1是同源域基因配对构象家族(Paired- Like Class of Homeodomain Genes)的成员。Hesx1的表达受Lhx3和PROP1的调控,Hesx1首先在脊索前板前体细胞内表达,胚胎发育第9.5天时在间脑和Rathke's囊表达,其在垂体的表达持续到发育第12天,Hesx1表达下调伴随PROP1转录增加,垂体细胞开始进行特异性分化。Hesx1基因敲除动物的表型不一致,5%的胚胎会发生垂体发育不全。Hesx1是抑制性转录因子,其N-末端区和同源域中各含有一个阻遏域,Hexs1同源域募集NcoR/Sin3/HDAC共阻遏复合物,N-末端eh1基序可与groucho相关的Tle共阻遏复合物相互作用,Hesx1/groucho(Tle)复合物的短暂调节在垂体器官形成过程中发挥重要作用 [7] 。
转录因子Rpx是Hesx1的同族体,其表达局限于口腔外胚层和Rathke's囊。多种抑制因子调控的Rpx/Hesx1表达衰减与腺垂体细胞终末分化标志物同时出现,提示Rpx/Hesx1基因表达下调在垂体发育过程的进展中发挥重要作用。Rpx可与Pro-1形成二聚体并抑制PROP1的活性,这提示Rpx/Hesx1可拮抗PROP1的生物学功能。
(4)成对同源域因子PROP1:PROP1是腺垂体细胞分化的决定性因子。PROP1 C-末端含有反式激活结构域,N-末端含有抑制结构域,这提示PROP1转录因子具有双向调节作用。PROP1/β-连环蛋白复合物激活Pit-1基因转录,而抑制Hesx1基因转录,PROP1转录激活/抑制作用的发挥取决于与其相互作用的辅助因子。PROP1表达起始时点与Rathke's囊闭锁的时点一致(E10.5d),表达峰值出现在胚胎第12.5天,随后,垂体细胞特异性分化终末期(E15.5~E16.5d)PROP1表达水平逐渐下调。Notch信号通路参与PROP1表达高峰的维持。PROP1基因缺陷鼠不表达Pit-1,垂体前叶各型激素分泌细胞群扩增障碍。PROP1基因突变导致多种垂体激素复合型缺乏。
(5)Six:发育中的垂体表达Six1、Six3、Six4、Six6基因。Six1和Six4基因共表达于很多胚胎原基结构,包括Rathke's囊;Six1或Six4基因缺陷的小鼠垂体可正常发育。Six3促进细胞增殖、拮抗Wnt信号途径和BMP通路,促进前叶垂体细胞特异分化。Six6在垂体早期发育阶段形成背-腹侧浓度梯度,胚胎第13.5天时表达降低。Six6基因敲除小鼠垂体和视网膜发育不全,机制与前体细胞分化障碍有关 [8] 。
(6)PAX6:PAX6在Rathke's囊背侧的短期表达参与垂体早期发育。在胚胎发育第9天,PAX6表达于口腔外胚层,和Shh表达区域无重合;胚胎发育第10~12天,Rathke's囊中可检测到PAX6表达并呈现出明显的背-腹浓度梯度;第13.5天,当垂体前体细胞分化开始时PAX6表达降低,直到第17.5天细胞分化进入终末期,PAX6在垂体中的表达消失。PAX6基因缺陷小鼠,垂体腹侧表达αGSU的细胞群(主要是促甲状腺激素细胞)增殖正常,而垂体背侧生长激素细胞和催乳素细胞数量显著减少。由此得出,PAX6决定垂体促甲状腺激素细胞/促性腺激素细胞、生长激素细胞/催乳素细胞的前体细胞的背-腹侧分区。
3.控制垂体各型细胞特异分化的转录因子:
Pit-1:Pit-1是一种POU同源异型结构域激素转录因子,是垂体特异的转录因子,其N-末端含有一个POU特异结构域,C-末端含有一个POU同源域,两个POU结构域中都含有高DNA亲和力的螺旋-转角-螺旋结构。Pit-1又被称为GHF-1,起初被发现有调节GH和PRL基因转录的功能;后期研究证实,Pit-1是3种特异化的垂体细胞(生长激素分泌细胞、催乳素分泌细胞、促甲状腺激素分泌细胞)增殖和分化的必要条件,并且抑制促性腺激素分泌细胞的分化。Pit-1依赖的细胞系和非Pit-1依赖的细胞系分别发育成促甲状腺激素分泌细胞和促性腺激素分泌细胞,这两种细胞系都来源于垂体腹侧,都表达糖蛋白激素共有的α亚基和激素特异的β亚基,Pit-1决定了垂体前体细胞的可塑性。胚胎第13.5天,Pit-1基因的表达需要PROP1和Wnt/β-连环蛋白信号通路的调节。
除此之外,Pit-1与GH、PRL、TSHβ亚基、GHRHR、1型生长抑素受体、TRH受体的编码基因启动子结合,与其他转录因子形成功能性异二聚体。Pit-1还能与甲状腺激素受体(Thyroid Hormone Receptor, THR)、视黄酸受体(Retinoic Acid Receptor, RAR)等核受体相互作用。锌指转录因子Gata2与Pit-1的相互作用对TSH分泌细胞的发育和TSH特异的β亚单位编码基因的表达有重要作用。
综上所述,复杂有序的信号机制精密调控下丘脑-垂体-甲状腺轴的发育,信号分子和转录因子间的相互作用介导包括促甲状腺激素分泌细胞在内的垂体细胞增殖、分化和区域化分布。
1.TSH细胞的发育:垂体前叶由Rathke's囊发育而来,小鼠胚胎第9.5天口腔外胚层中部凹陷使发育中的下丘脑和垂体直接相连。垂体器官形成过程包括祖细胞在一系列调节信号的作用下增殖、分化、终分化的过程,如BMP4、FGF8、Wnt/β-连环蛋白、Notch等多重信号通路的调控作用使特定的转录因子时间及空间特异性表达,这些转录因子早期参与细胞系形成、形态及功能分化,后期参与激素合成基因的转录调节。胚胎第11.5~13.5天,大部分垂体细胞终止细胞增殖,结构与功能相近的细胞群迁移至垂体的特定区域形成不同的功能区,负责整合及传导生理信号刺激。
糖蛋白激素α亚基(αGSU)是垂体在发育过程中最早表达的激素合成基因(约在胚胎发育第10.5天),毗邻神经上皮表达的Wnt5a和BMP4是最早期的调节信号,继而Hesx1、Ptx1/2和Lhx3/4开始表达。胚胎发育第12.5天时腹-背侧BMP梯度和背-腹侧FGF梯度的形成促进了TSHβ亚单位分泌细胞在垂体喙部尖端初步形态的形成,至出生时喙部尖端TSHβ亚单位分泌细胞消失;而胚胎第15.5天时,在垂体特异的转录因子Pou1f1(Pit-1)的调节作用下,垂体体部TSHβ亚单位分泌细胞群逐渐形成,这个区域Pit-1和TSHβ的表达是持续存在的。锌指转录因子Gata2在TSH分泌细胞的分化过程中发挥重要作用,Gata2于胚胎第10.5天时在垂体前叶表达,它与TSHα基因启动子结合并反式激活TSHα基因,Gata2与Pit-1协同作用促进TSHβ基因的表达。Gata2基因敲除小鼠TSH分泌细胞数量和血清TSH、FSH水平较野生型降低,说明Gata2在维持正常的甲状腺功能和性腺功能中至关重要 [9] 。胚胎发育早期,几乎所有的垂体细胞都表达干细胞标记分子Sox2;成人垂体中存在表达Sox2和巢蛋白(Nectin)多功能干细胞群,这类干细胞具有分化为成熟垂体细胞的潜能,可能在机体需求波动时对维持正常的垂体功能有重要作用,也可能进展为垂体肿瘤。
2.TSH细胞的解剖:TSH细胞约占垂体前叶细胞总数的5%,主要聚集于垂体前叶的前内侧(图4-2-1),其形态小于其他细胞类型,形状不规则且有细长的胞质突起(图4-2-2),细胞核呈圆形或卵圆形,胞浆内富含PAS-/AF-/AT+的分泌颗粒(图4-2-3)。免疫组织化学方法是光学显微镜下观察TSH细胞最可靠的方法,它不易被酸性染料和苏木精-伊红染色,相比于垂体促肾上腺激素细胞碱性染料和PAS着色浅。电镜下,促甲状腺细胞胞浆丰富,卵圆形细胞核位于细胞中央,粗面内质网上散在着很多囊泡。高尔基复合体聚集在核周,其形态随激素合成活性而变化。棒状结构的线粒体在电镜下呈电子致密团样结构,内部可见规则排布的横嵴。吞噬溶酶体多见。小球状的分泌颗粒直径为100nm至200nm不等,电子密度较小,分泌颗粒胞吐并不可见。
TSH分泌细胞电镜结构:胞浆内富含分泌颗粒 [10] 。
3.TSH细胞的细胞学:免疫电镜证实,人促甲状腺激素细胞的分泌颗粒中富含TSH;免疫组织化学染色显示,TSHα亚单位和β亚单位呈现胞浆强阳性,而并没有检测到其他激素的免疫活性。而在大鼠垂体中存在同时表达TSH和GH的细胞,提示生长激素细胞和促甲状腺细胞在功能上的密切联系。胚胎发育过程中,生长激素细胞与促甲状腺细胞都来源于表达Pit-1转录因子的祖细胞。有研究报道,在丙硫氧嘧啶处理的大鼠甲减模型中,垂体生长激素细胞发生脱颗粒样变化并向促甲状腺细胞转化。在长期患有原发性甲状腺功能减退的患者的垂体中,也可以发生生长激素细胞向促甲状腺细胞的反向分化。以上研究提示,不同类型的腺垂体细胞并不能被完全分割开,在特定条件下,一种细胞可向另一种细胞类型转化并发挥相应的激素分泌功能。因此,一直被大家所公认的“一种细胞,一种激素”的说法或许不再科学。
促甲状腺细胞的存在持续终生,其数量、分布、形态特点及超微结构不存在性别间的差异,促甲状腺细胞不随年龄的增长发生退化,可以持续地分泌TSH;实际上,老年人的TSH水平较青、中年人群TSH水平升高,亚临床型和原发性临床型甲状腺功能减退的发病率在老年人群高于青、中年人群。

图4-2-1 TSH分泌细胞在垂体的分布:TSH分泌细胞约占垂体前叶细胞总数的5%,主要聚集于垂体前叶的前内侧
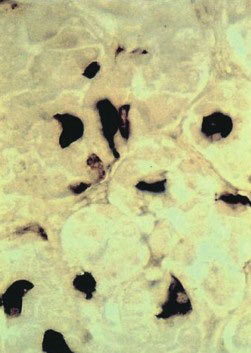
图4-2-2 TSH分泌细胞的形态:TSH分泌细胞呈角状
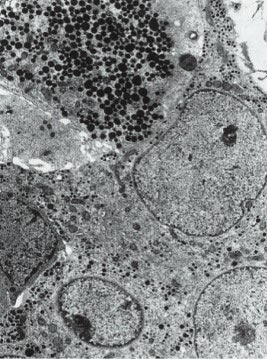
图4-2-3 TSH分泌细胞电镜结构:胞浆内富含分泌颗粒
TSH是一种糖蛋白激素,由非共价结合的α亚单位、β亚单位构成异二聚体,其中α亚单位为TSH、FSH、LH和CG共有,β亚单位决定TSH的特异生物学功能。TSH分子中由3个二硫键在中央结点处形成两个发夹环样结构,每个亚单位的基序结都位于2个发夹结构环(L1和L3)的侧翼区,发夹环位于基序结的N-末端,TSH分子的C-末端有一个双股β结构类似薄板的长环(L2)。β亚单位的结构环缠绕着α亚单位形成了一个安全带样的结构固定住α亚单位。
α亚单位和β亚单位由存在于不同染色体上的不同基因编码。TSH分泌细胞内存在特定的转录因子与基因调节区结合并与泛素化因子相互作用调节基因转录过程。经大量的生物化学研究证实,TSH分泌细胞内α亚单位和β亚单位基因的激活或抑制,从根本上取决于核染色质的修饰状态,包括组蛋白修饰和DNA甲基化状态的改变。当接收到TRH等刺激性生理信号或T 3 等抑制性生理信号时,转录因子与基因启动子区域结合并招募染色质修饰酶,从而启动基因转录或导致基因沉默。
1.TSHβ亚单位基因:TSHβ基因是含有4527个碱基对的单拷贝基因,位于1号染色体短臂q13.2位点,基因结构包含3个外显子和2个内含子。后来,在人类垂体、白细胞、甲状腺中发现低水平的含有编码序列的TSHβ mRNA的剪切异构体,并在血清中分离出其相应的TSH β肽段,其病理生理学作用有待进一步研究证实 [11] 。TSH β基因启动子区域靠近转录起始位点处存在很多启动转录和调控基因表达的元件,如图4-3-1所示,被大家所熟知的TATA框是位于转录起始位点上游28bp处对于定位RNA合成位置至关重要的顺式作用元件。将小鼠TSHβ基因启动子区5 ′ 端敲除并与荧光素酶报告系统连接的方法明确了TSHβ基因启动子区域270bp位点处基因表达所需的顺式作用元件的序列(图4-3-2);而这仅仅明确了很短的一段基因的序列,启动子上游6kb处的位点也和基因表达有关系,具体序列仍不得而知。
启动子缺失研究证明,小鼠TSHβ基因启动子-271~-80区段足以使TSH分泌细胞具有特异的生物学活性,并且TSH分泌细胞内的转录因子可以结合到近侧的启动子上。用核蛋白提取的方法,在这段较长的区段中发现了4个与蛋白交互作用的位点。同源结构域因子Pit-1和锌指转录因子Gata2都可以与TSHβ基因启动子-135~-18区段的序列结合;两个转录因子都可以独立地结合到启动子区,与DNA形成异聚体复合物,两者在结构上相互调整,在功能上协同促进TSHβ基因启动子活化。也有研究提示,下游-88位点附近的抑制序列抑制Gata2的转录激活作用,而Pit-1可解除对Gata2活性抑制。
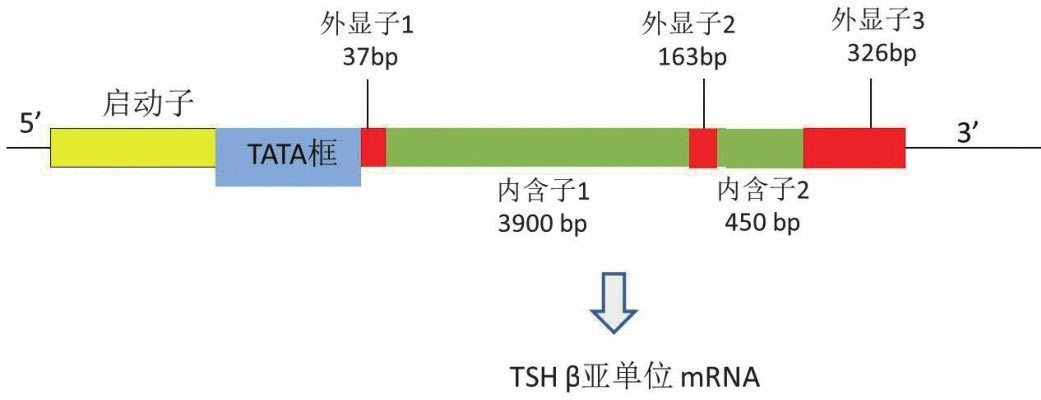
图4-3-1 TSHβ亚单位基因内含子及外显子的相对位置和大小,对RNA转录起始点定位至关重要的TATA框结构靠近第一个外显子上游
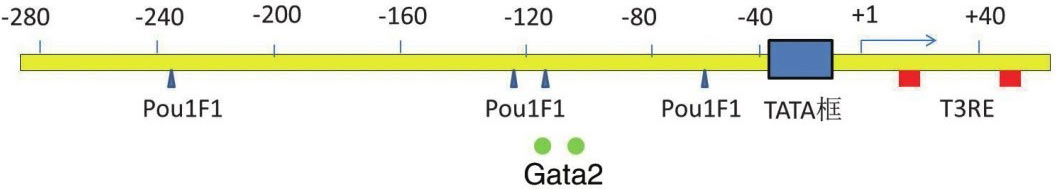
图4-3-2 TSHβ亚单位基因启动子区重要的DNA结合元件Pou1F1、Gata2的及T 3 反应元件的结合位置
转录因子TRAP220 (MED1)募集到启动子处与转录启动有关。起初,TRAP220被认为是与配体化的甲状腺激素受体相互作用的转录介质复合物的一部分。TRAP220单拷贝基因小鼠垂体TSHβ基因转录水平降低并伴发甲减。因TRAP220不能直接与DNA结合,它依靠与Pit-1、Gata2在结构上的嵌合性募集到TSH β基因。非垂体细胞单独转染Pit-1、Gata2、TRAP220并不会引起TSHβ基因的表达变化,而当三者共转染时,可在最大限度上促进TSHβ基因的表达。蛋白交互作用实验证实,在体内和体外条件下,Pit-1、Gata2、TRAP220这3个转录因子之间都可以发生相互作用,介导因子间相互作用的结构域对功能最大化起到重要作用 [12] 。因此,TSH分泌细胞内TSHβ基因的表达受到一系列转录因子的共同调控,或直接作用于启动子,或通过蛋白-蛋白相互作用促进基因转录激活。
2.TSHα亚单位基因:编码人类糖蛋白激素α亚单位的基因位于6号染色体6q12-q21,长度为9635kb的单拷贝基因,基因结构包括4个外显子和3个内含子,转录起始位点上游26bp处有TATA框。
α亚单位基因在TSH分泌细胞、促性腺细胞、胎盘细胞都有表达,但基因表达调控具有细胞特异性。不同细胞类型的特异表达取决于启动子的不同区域,下游启动子(-200bp)调控胎盘细胞内的表达;中间区(-225~-200bp)与类固醇生成因子1(Steroidogenic Factor 1)结合从而调控促性腺细胞的表达;上游序列对调控TSH分泌细胞内的表达至关重要,从-342bp延伸至-329bp的垂体糖蛋白激素基础序列可与P-Lim结合,对调控TSH分泌细胞和促性腺细胞的基因表达有重要作用。-480~-300bp区段的序列可能与小鼠TSH分泌细胞的α亚单位基因表达有关,而与促性腺细胞基因表达无关,其中-434~-421bp的序列可以和同源结构域的转录因子Msx1相互作用。启动子区转基因小鼠实验表明,上游-4.6~-3.7kb的DNA基序中存在Gata、SF1、ETS等多个转录因子的结合位点,即参与TSH分泌细胞的基因调控,也可调节促性腺细胞的基因表达,这提示我们转录因子结合毗邻的顺式作用元件与远端增强子之间的协同效应 [13] 。
完整的TSH分子是由非共价结合的α亚单位和β亚单位构成的分子量为28kDa的异质二聚体。其中α亚单位为TSH、FSH、LH和CG共有,含有92个氨基酸,TSH分子特异的β亚单位由118个氨基酸组成。TSH分子的合成过程中受多种因子和信号的精密调控,包括转录和翻译、糖基化、折叠、亚基结合等步骤。
1.转录和翻译:TSH β亚单位和α亚单位基因(图4-4)在一系列特异的转录因子调控下转录为RNA前体,RNA前体在内含子与外显子接点处经过精密的剪切过程形成成熟的mRNA分子,并从细胞核转运到细胞质进行遗传密码的翻译。正常情况下,TSHα亚单位和β亚单位基因的转录在一系列生理性信号的调控下得以协调地进行,其中TRH和T 3 是最主要的生理性调节因素。核糖体携带TSHα亚单位和β亚单位的mRNA并各自转运到胞浆,TSHβ前20位氨基酸和TSHα前24位氨基酸各自构成“信号肽”,疏水性的信号肽可以插入到粗面内质网的磷脂双分子层内,TSH β亚基和α亚基原始肽的合成在粗面内质网的内腔中进行,翻译过程完成前信号肽段被剪切掉形成成熟的TSH β亚基和α亚基,最终形成的TSH β亚基含有118个氨基酸、α亚基含有92个氨基酸序列。
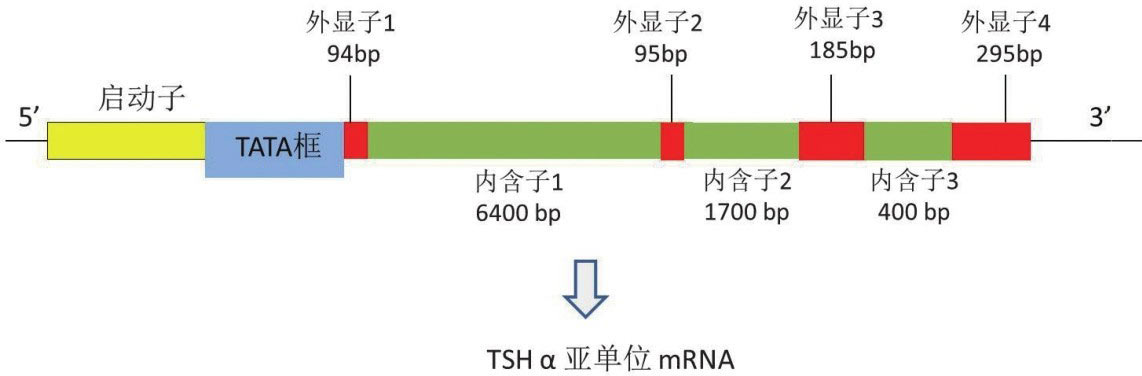
图4-4 TSHα亚单位含因内含子及外显子的相对位置和大小,对RNA转录起始点定位至关重要的TATA框结构靠近第一个外显子上游
2.糖基化:糖基化修饰过程使TSH亚单位的生物学活性发生改变,TSHβ亚单位只在第23位天冬氨酸残基处有一个糖基化修饰位点,而α亚单位可分别在52位和78位天冬氨酸残基处糖基化修饰。细胞内过多的游离α亚基还会在第39位苏氨酸残基上发生糖基化修饰,这个位点被认为与α亚基和β亚基的结合有关,究竟是苏氨酸残基糖基化修饰抑制了α亚基和β亚基的结合还是因为游离α亚基氨基酸残基的暴露引发了翻译后修饰仍有待进一步研究证实。很多学者对TSH亚基翻译后糖基化修饰的过程进行了深入的研究,第一步是将含有14个残基(2个葡萄糖残基-9个甘露糖残基-3个N-乙酰葡糖胺)的寡聚糖组装到磷酸多萜醇载体上,然后在寡糖转移酶的辅助下转移到TSH亚基的天冬氨酸残基上,寡糖转移酶可以识别天冬氨酸-X-丝/苏氨酸三肽序列。14糖基寡聚糖在内质网腔和高尔基复合体中逐步被裂解,生成一个只含有6个糖基的中间产物,随后N-乙酰葡糖胺、半乳糖、N-乙酰半乳糖胺整合到6糖基中间产物上形成了一个复杂的寡糖分子,N-乙酰半乳糖胺的硫化作用和半乳糖残基的唾液酸化(Sialation)也相继在远侧高尔基体中发生 [14] 。硫化作用增加TSH分子的生物学活性,而唾液酸化过程降低其生物学活性。
3.折叠:通过X线晶体学分析法得到的CG分子的三维结构为我们构建TSH分子结构提供了模型,TSH分子中由3个二硫键在中央结点侧形成两个发夹环样结构,每个亚单位的基序结都位于2个发夹结构环(L1和L3)的侧翼区,发夹环位于基序结的N-末端,TSH分子的C-末端有一个双股β结构类似薄板的长环(L2)。β亚单位的结构环缠绕着α亚单位形成了一个安全带样的结构固定住α亚单位。肽链的折叠过程在转录结束前就已经开始,折叠过程依赖于氨基酸残基的糖基化修饰,肽链的折叠使分子内部的二硫键结构得以维持蛋白质的三维结构,有利于α亚基和β亚基的结合。
4.亚基结合:肽链合成、折叠以后,TSHα亚单位和β亚单位的结合很快在内质网腔中开始,至高尔基复合体中完成结合过程。两个亚基的结合促进了α亚基的寡糖修饰过程。TSHβ亚基第27~31位氨基酸序列(CAGYC)在不同物种间是高度保守的,被认为与α亚基的结合有关。CAGYC基因区点突变可致先天性甲状腺功能减退,机制在于突变基因合成的TSHβ亚基不能与α亚基结合,不能合成完整的具有生物活性的TSH分子。β亚基必须与α亚基一起分泌到运输小泡中,不存在游离的TSHβ亚基。基因突变所致的TSHβ亚基第25位丝氨酸糖基化识别位点的苏氨酸替代会使TSH分子的合成效率降低70%,可能与邻近的CAGYC区结构破坏有关,而苏氨酸替代后的糖基化位点并没有发生变化 [15] 。
TSH分子和游离α亚基在高尔基复合体合成加工后被转运到分泌小泡,分泌过程主要受TRH和其他下丘脑激素的调控。
健康成年人的TSH生成率为每天100~200 mU,血液循环中的半衰期约为50min,清除效率为50mL/min。甲减患者的TSH分泌效率可比正常水平高出10~15倍,而清除率会稍微降低;甲亢患者的TSH分泌受到抑制而清除代谢过程加快 [16] 。
1.TSH发生学:人胚胎发育8~10周时,下丘脑中可检测到TRH,至妊娠末期TRH水平逐渐升高。第12周时,胚胎垂体和血清中可检测到具有免疫活性的TSH但维持在很低的水平,直到18周血清和垂体中的TSH迅速上升,血清T 3 、T 4 水平也相继升高。妊娠20~40周,胎儿血清TSH和T 4 持续升高。垂体在妊娠晚期就可以接受外源性TRH的刺激而分泌TSH,而TSH的负反馈控制直到出生后1~2个月才建立起来。
足月儿出生后30min内血清TSH水平骤然升高,可能与寒冷刺激和宫外环境变化有关,出生后3~5d回落到成人水平。出生后4h内血清T 3 水平升高,出生后24~36h后血清T 4 水平也有小幅度增加,1~2个月后达到稳定水平。早产儿血清TSH水平变异较大,出生时通常低于足月儿血清TSH水平,出生后1周内略减低,随后逐渐升高至正常水平。患有其他疾病的早产儿出生时血清TSH更低,在恢复的过程中逐渐升高至正常水平。
2.TSH分泌模式:类似其他垂体激素,TSH有基础分泌和脉冲分泌两种分泌模式,基础分泌占循环TSH总量的40%~50%,脉冲分泌占50%~60%。如图4-5所示,通常24h内有15~20次TSH脉冲峰,每次平均持续90min。血游离T 3 峰值在TSH脉冲峰90min后出现,说明脉冲分泌的TSH是促进甲状腺分泌T 3 的直接因素。但游离T 3 的上下界值差只占平均游离T 3 水平的11%左右,可能因其在血清中的半衰期较长,而且大部分游离T 3 不是直接来源于甲状腺。TSH的分泌存在明显的昼夜节律,夜间活动水平较白昼升高2倍。在有正常睡眠周期的个体,TSH在23时至次日5时处于最高水平,白日峰值出现在9时至12时。4周龄以下的新生儿TSH分泌不存在昼夜节律,至出生后1~2个月逐渐形成,在健康儿童中TSH分泌的昼夜节律已经正常建立。
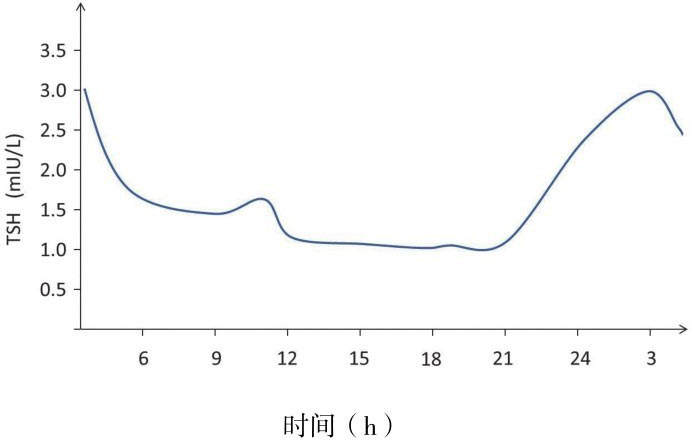
图4-5 健康成年人TSH分泌昼夜节律:一般在23时至次日5时处于最高水平,白日峰值出现在9时至12时
TSH水平的昼夜变化是夜间脉冲分泌量增加的结果。与其他垂体激素不同,TSH的夜间分泌高峰是非睡眠依赖型的,夜间TSH水平升高可在睡眠前发生,睡眠剥夺可使TSH分泌增加。原发性甲状腺功能减退的患者仍存在脉冲分泌和昼夜分泌节律,脉冲分泌频率与甲状腺功能正常者相同,而基础分泌和脉冲分泌水平都显著升高并随甲减程度的增加进一步升高。在患有严重的原发性甲状腺功能减退的患者,TSH分泌曲线的振幅大大增加并且在白昼持续出现,TSH水平昼夜变化规律因而变得不明显但仍然存在,左甲状腺素(L-T4)治疗可使TSH分泌节律恢复正常。相比而言,下丘脑-垂体因素导致的甲状腺功能减退TSH分泌水平降低、夜间TSH脉冲峰消失。其他危重病状态时也会出现TSH分泌水平降低和节律消失。
TSH脉冲分泌的起源尚不清楚。甲状腺激素改变TSH脉冲振幅但不影响脉冲频率 [17] 。目前推测TSH分泌节律的调控器存在于下丘脑,TRH神经元和其他神经-内分泌神经元协同作用刺激垂体的TSH脉冲分泌。然而超生理剂量的TRH输入并不改变人的TSH脉冲分泌频率。生长抑素和多巴胺都可降低TSH脉冲分泌的幅度,同样不影响其频率。目前唯一报道的TSH脉冲频率发生改变的临床状况是垂体瘤,24h内出现27次TSH脉冲峰。大鼠垂体前叶的2型脱碘酶活性会出现昼夜变化,可能参与调节TSH的昼夜分泌。
生理情况下,血皮质醇可能是控制TSH分泌昼夜节律的重要因素,虽然皮质醇并不影响其脉冲频率。皮质醇功能减退的患者停用糖皮质激素后,日间TSH水平升高,TSH昼夜节律消失。模仿生理性的皮质醇分泌模式给予这些患者生理剂量的氢化可的松以后,昼间TSH水平降低,正常的TSH昼夜节律重新建立;同等剂量的氢化可的松24h内常量泵入也可降低TSH水平,但TSH分泌并无昼夜节律。健康成人接受内源性皮质醇合成抑制剂美替拉酮(Metyrapone)治疗后,也会出现TSH水平升高和昼夜节律消失 [18] 。以上表明,内源性血皮质醇的早高峰使血清TSH水平短暂降低,从而形成其昼间水平的变化规律。
中枢神经系统的信号分子和外周循环的反馈因素共同调节TSH的生物合成过程。下丘脑TRH是促进TSH生物合成的最主要分子,循环中的甲状腺激素水平是TSH最强有力的负性调节因素,其他下丘脑激素和外周循环激素分子也参与调节TSH合成过程。
1.TSHβ亚单位合成的下丘脑调节:促甲状腺激素释放激素(Thyrotropin-Releasing Hormone , TRH)是下丘脑室旁核合成的三肽激素,经下丘脑-垂体门脉系统运输到垂体,可使编码TSHα亚基和β亚基的基因转录水平上调3~5倍。TRH的转录和翻译后修饰过程都受到T 3 的抑制性调节。母体或胎儿来源的TRH不参与垂体促甲状腺细胞的发生、发育,TRH基因缺陷小鼠出生时不发生甲减,TRH参与出生后的TSH活性维持 [19] 。
TRH与垂体促甲状腺细胞上的特异膜受体TRHR1结合后启动细胞内信号通路级联反应,TRHR1基因敲除可引发中枢性甲减。在大鼠垂体瘤GH3细胞系中,TRH-受体复合体与质膜内侧的G蛋白(G)相互作用后结合并激活鸟嘌呤核苷酸(G ' ),G ' 与磷脂酶C(C)结合并将其激活(C ' ),C ' 催化4,5-二磷酸磷脂酰肌醇水解成两个细胞内第二信使分子:三磷酸肌醇(Inositol Triphosphate)和1,2-二酰基甘油(1,2-Diacylglycerol)。三磷酸肌醇引起内质网Ca 2+ 释放,从而激活分泌颗粒向细胞膜转移并以胞吐形式完成TSH分泌。与此同时,1,2-二酰基甘油诱发的磷脂酶C激活使细胞内一系列蛋白发生磷酸化而促进胞吐分泌。在垂体前叶和GH3细胞系中,TRH可激活核蛋白Islet-brain-1 (IB1)/JIP-1,被认为与增强促甲状腺细胞TSHβ基因表达有关。TRH促进催乳素细胞分泌催乳素也有磷脂酰肌醇-蛋白激酶C-Ca 2+ 通路的参与。而TRH对TSHβ基因启动子区域的作用可能由转录因子AP-1介导。
TSHβ基因启动子-128~-61bp区段和-28~+8bp区段是TRH的两个反应区,其上游区域含有垂体特异性转录因子Pou1f1的结合位点,这提示TRH调节TSHβ基因表达过程有Pou1f1的参与。大鼠TSHβ基因的TRH反应区定位在上游-204位点附近。有研究证实,在佛波酯(Phorbol)和cAMP的作用下,PKC或PKA途径介导的Pou1f1磷酸化可使Pou1f1与TSHβ基因的反式作用元件结合发生改变 [20] 。非垂体细胞过继转移研究表明,TRH增强Gata2介导的TSHβ基因启动子激活效应;Gata2锌指结构域改变或TSHβ基因启动子Gata2反应元件发生突变都会导致TRH不能发挥其生物学作用。其他转录活化因子如TRAP220/MED1等,也参与TRH对TSHβ基因的调控。
多巴胺通过激活DA2多巴胺受体抑制TSHα亚单位和β亚单位基因的转录,机制与细胞内cAMP水平降低有关。TSHβ基因启动子-128~-61bp区段和+3~+8bp区段是cAMP发挥作用的必要位置。上游区域与TRH反应区域一致,也包含Pou1f1结合位点,下游区域被T 3 反应区(+3~+37)和TRH反应区(-28~+8)包括,下游区域与AP-1结合区(-1~+6)有交叉重叠。-1~+6区段的基因序列与Pou1f1共同作用介导cAMP和TRH对基因表达的调控。由此可见,转录因子和激素性调节因子的多元互动效应在基因启动子区域的聚集使得TSH生物合成的精密调控得以实现。
2.TSHβ亚单位合成的外周调节:
(1)甲状腺激素抑制TSHβ亚单位合成的生物学过程:甲状腺激素主要通过细胞核内的甲状腺激素受体(TR)发挥作用,甲状腺激素通过多种膜转运蛋白进入细胞内继而弥散到细胞核内激活下游信号通路。T 4 是几乎不具有生物活性的激素原,是组织内的碘化甲状腺原氨酸脱碘酶家族的反应底物。膜结合的同型二聚体硒蛋白酶以时间和组织特异性特点激活T 4 (主要与1型和2型脱碘酶有关)或灭活T 4 (主要由3型脱碘酶介导) [21] 。2型脱碘酶(D2)是激活T 4 的主要脱碘酶,与底物有很高的亲和力,它存在于核周的内质网上,可将T 4 转化为T 3 ;在T 4 存在的条件下,2型脱碘酶活性在泛素蛋白酶体的调控下很快消失。大鼠垂体促甲状腺素细胞表达2型脱碘酶mRNA和蛋白,甲减状态下表达增加。鼠类TtT-97肿瘤的促甲状腺素细胞中高水平表达2型脱碘酶,即使在T 4 超生理量的状态下也可以持续产生T 3 。在给予T 3 或T 4 后,正常小鼠的血清TSH水平会降低,而当目的性敲除D2基因或给予D2抑制剂胺碘酮时,只有T 3 可以发挥对TSH的抑制效应。由此可见,2型脱碘酶在控制甲状腺激素对TSH的反馈调节中发挥至关重要的作用。
除经典核受体途径以外,甲状腺激素在细胞膜或细胞质的非基因组作用也参与某些基因的基础转录效率设定或细胞活动的调控。T 4 或高水平T 3 可以与细胞表面的αv/β3整合素受体结合,继而活化丝裂原激活化蛋白激酶(Mitogen-Activated Protein Kinase, MAPK)信号通路级联反应,引发细胞内或核内一系列生物学效应。T 3 可激活磷脂酰肌醇-3-激酶(Phosphatidylinositol 3-kinase, PI3K),参与TRα在胞浆和胞核间的穿梭运动、特定基因的转录调控、Na + -K + -ATP酶向胞膜的植入等。细胞质中的T 3 可直接激活PI3K/Akt信号传导途径。有些甲状腺激素还可在细胞器内(如线粒体)发挥作用,影响线粒体发生、复制和能量产生 [22] 。甲状腺激素的很多非基因组学效应可能对调节某些基因的基础转录效率和细胞内生化反应起重要作用。
动物模型及垂体瘤细胞实验证实,T 3 可显著降低TSHα亚单位和β亚单位基因的转录效率,T 3 对TtT-97细胞系TSHα亚单位和β亚单位基因的转录抑制在30min时已经非常显著,4h达到最大程度;在蛋白合成抑制剂环己酰亚胺(Cycloheximide)存在的条件下,这种抑制效应仍然存在,说明T 3 对TSH基因的抑制作用不需要中间介质蛋白。T 3 与其核受体结合后主要在转录水平调控基因表达,调控效应取决于核受体与T 3 的结合比率,T 3 核绑定与转录抑制过程在时间进程上保持一致。
(2)甲状腺激素抑制TSHβ亚单位合成的分子机制:甲状腺激素在不同的细胞类型中激活不同的信号传导途径,主要参与元素有:甲状腺激素转运蛋白,TR多个受体亚型,配体占用,受体二聚化,TR反应元件在目的基因上的结合位点,辅助活化/抑制因子。此外,在代谢调节和发育过程中,甲状腺激素相关信号通路分子可以加强其他信号通路的生物学效应。T 3 对TSH的抑制效应需要与TR结合,主要是TRβ1、β2亚型,因为在甲状腺激素抵抗和TSH不适当分泌的患者中大多是TRβ基因突变,近期TRα基因突变也有报道。TRβ与转录起始位点附近的特定顺式激活DNA序列结合,TSHβ基因的T 3 反应元件位于+3~+37区段,+3~+13、+28~+37两个T 3 受体结合位置介导T 3 对TSH的抑制效应。T 3 反应可通过受体单体、同源二聚体发挥,也可通过异源二聚体类视黄醇-X受体(Retinoid-X Receptors, RXR)发挥。TtT-97促甲状腺素肿瘤细胞系和TαT 1 促甲状腺细胞系体外实验证实,RXR的选择性配体抑制TSHβ基因的表达,这个现象已在肿瘤患者得到验证,给予维甲酸贝沙罗汀(Retinoid Bexarotene)治疗后诱发中枢性甲减(低T 4 伴低TSH)。RXR选择性视黄酸LG 268,降低小鼠血循环中的TSH和T 4 水平,同时伴有TSHβ基因mRNA表达降低,而对TRH无影响。以上提示其对垂体促甲状腺细胞的直接抑制作用。
也有研究对人TSHβ基因外显子1附近的负性反应元件的功能提出异议,因为将其敲除后并不削弱T 3 对TSHβ基因启动子活性的抑制效应。配体化的TRβ可通过其DNA结合域与Gata2的锌指结构域相互作用。结合T 3 的TR可与TRAP220相互作用。因此,可能的机制是,具有转录激活功能的Pou1f1/Gata2/TRAP220复合体与TSHβ基因启动子的干涉作用在介导T 3 对TSH合成的负反馈效应中至关重要。
T 3 可通过很多机制促进基因转录。在配体不存在的情况下,TR与DNA顺式激活反应元件结合,与核受体辅阻遏分子家族相互作用,募集组蛋白去乙酰化酶进行染色质结构修饰,从而导致目的基因的表达抑制。当T 3 存在时,核受体辅阻遏分子很快与TR解离,取而代之的是转录辅助复合体与TR结合,增加局部组蛋白甲基化水平和乙酰化水平,使染色质结构松散而利于基因转录。有激活功能的转录因子,如TRAP220随后募集到TR,通过蛋白间相互作用而激活Ⅱ型RNA聚合酶介导的基因转录。
相比而言,T 3 对TSH亚单位基因的负性调控机制并不是很清楚。与配体结合的TRβ被报道可募集组蛋白去乙酰化酶3,降低H4组蛋白的乙酰化水平,从而使TSHβ基因结构紧密不利于转录起始。在无T 3 作用条件下,NcoR/SMRT、TBL1、HDAC3组成的辅阻遏复合物和TRβ结合,共同作用于TSHα基因启动子,决定组蛋白乙酰化水平和基因转录基础效率。T 3 与TR的结合使NcoR/SMRT、TBL1、HDAC3辅阻遏复合物与TR解离,致使某些基因位点的组蛋白H3乙酰化水平升高,cAMP引起的H4乙酰化作用使TSHα基因被负性调控 [23] 。多项体内及体外研究证实,TSHβ基因的负性调控需要TRβ完整的DNA结合域的存在。在一项研究中发现,联用Pou1f1和Gata2可以激活同时含有TRβ1基因序列的质粒重组DNA中的TSHβ(-128/+37)受体基因,无论T 3 存在与否。研究者还发现,未与T 3 结合的TRβ不能增强启动子活性,而缺失N-末端和DNA结构域的TR β1分子不能传达T 3 刺激对TSHβ基因启动子的负性调控 [24] 。转基因小鼠模型中,利用基因靶向技术将缺乏DNA结构域的TR β基因取代野生型基因并不改变配体和辅助因子之间的相互作用。TR β纯合子突变的小鼠会发生中枢性甲状腺激素抵抗,血清TSH水平较对照小鼠升高20倍伴血T 3 2~3倍升高,这种表现型与TR β敲除的小鼠相同。
促甲状腺细胞含有甲状腺激素受体的所有亚型:TR α1、TR β1、TR β2和非T 3 结合的变构体。垂体和下丘脑内的T 3 反应性TRH神经元主要表达TR β2,对TSH的调节至关重要。TR β2基因敲除鼠表型与垂体型甲状腺激素抵抗一致,即使在TR β1和TR α1存在的情况下,血清TSH水平和甲状腺激素水平显著升高,提示TR各异构体在功能上不存在代偿机制。TR β而不是TRα可与TSH β基因启动子区相互作用;TR β基因敲除而TRα正常表达的细胞中,T 3 对TSHβ基因转录的抑制效应完全消失TR和TR辅助蛋白组成的异二聚体,例如RXR,也可以与DNA结合形成异二聚体复合物,它们与特定DNA序列结合的亲和力各不相同,功能活性也各不相同 [25] 。RXR γ1是RXR的一个亚型,只在促甲状腺素细胞表达,可通过启动子区-200~-149区段的序列介导9-顺式维甲酸的对小鼠TSHβ基因的抑制作用,这段位于上游的序列不同于介导甲状腺激素转录抑制作用的常见序列。其他与TR相互作用的蛋白分子还包括共激活因子,如糖皮质激素受体相互作用蛋白-1 (Glucocorticoid Receptor Interacting Protein-1, GRIP-1)、类固醇受体共激活因子-1(Steroid Receptor Coactivator-1, SRC-1);辅抑制因子,如甲状腺激素受体和视黄酸受体的沉默中介蛋白(Silencing Mediator)、核受体辅阻遏因子(Nuclear Receptor Corepressor , NCoR)。
很多研究聚焦于NCoR1与TRβ相互作用介导的TSH亚单位基因负性调控的分子机制。转基因小鼠模型表明,辅阻遏因子NCoR1对下丘脑-垂体-甲状腺轴的生理学作用是由垂体促甲状腺细胞表达的缺乏TR相互作用结构域的蛋白突变体完成的。虽然NCoR1突变小鼠的T 3 、T 4 水平较对照鼠降低,而血清TSH水平并不改变,表型与中枢性甲减一致;这项实验的研究结果也证实了低水平T 3 条件下,NCoR1对TSH亚单位基因表达的激活作用。在其他中枢性甲减的动物模型中,TSH水平轻度升高,而下丘脑-垂体-甲状腺轴无反应。以上结论提示,NCoR1可通过调节甲状腺激素受体介导的TSH抑制效应而重新设定甲状腺轴并控制垂体TSH分泌。
甲状腺激素可同时从下丘脑和垂体水平调节TSH分泌,有很多研究致力于评估两者的相对作用效果。应用TRH或TR β单基因敲除鼠,或两者同时敲除的小鼠,结果证实,TRH在体内下丘脑-垂体-甲状腺轴的调节中发挥主要作用,下丘脑分泌TRH的细胞是TSH合成和甲状腺激素合成的必要条件,并且控制下丘脑-垂体-甲状腺轴的调定点。甲减时,促甲状腺素细胞对反馈调节信号做出的适当的反应需要TRβ和TRH的共同参与。
3.TSHα亚单位合成的下丘脑调节:下丘脑来源的TRH调节TSHα亚单位的合成需要两个转录因子P-Lim和CREB结合蛋白(CBP)的参与。CBP可与TSHα亚单位基因启动子-151~-135区段结合,Pou1f1样蛋白可与远端-223~-190区段结合,人糖蛋白激素α亚单位基因启动子区的CRE元件由-146~-111区段的长度为18bp的重复序列组成。TRH刺激P-Lim和CBP的转录,两个转录因子间的相互作用协同参与α亚单位基因启动子的激活。P-Lim直接与TSHα亚单位基因启动子区结合,而CBP不直接与DNA结合,它必须借助转录因子间的相互作用募集到启动子区,而且只有在TRH刺激存在的条件下才能形成P-Lim/CBP复合体。在转染过表达的非垂体CV1细胞系,TRHR1的表达使Gata2依赖的TSHα基因启动子转录激活增加,这表明TRH信号通路有Gata2的参与,可同时激活TSHβ和α亚单位的基因表达。多巴胺可同时抑制促甲状腺细胞TSHα基因和β基因的表达,机制与细胞内cAMP水平降低有关 [26] 。
4.TSHα亚单位合成的外周调节:甲状腺激素抑制促甲状腺细胞TSHβ亚单位基因转录的同时也抑制α亚单位基因转录。人糖蛋白激素α亚单位基因启动子区的T 3 反应元件定位于-22~-7区段,在啮齿类动物高度保守。T 3 受体的不同亚型和辅助抑制因子SMRT、NCoR的相互作用介导T 3 的抑制效应。有研究证实,α亚单位基因启动子区T 3 反应元件突变可导致其不能被TR结合,但并不会消除T 3 对基因转录的抑制作用,提示在T 3 抑制α亚单位基因表达的过程中蛋白-蛋白相互作用比蛋白-基因相互作用更重要。
5.TSH糖基化调节:TRH和甲状腺激素是参与TSH分子糖基化修饰过程的主要调节分子。原发性甲减、甲状腺激素抵抗或TRH治疗等可通过提高TSH分子的糖基化水平而使TSH生物学活性升高。中枢性甲减、TSH分泌性垂体瘤、甲功正常的尿毒症等疾病状态时,TSH分子糖基化水平都会发生变化,低聚糖残基的硫化作用和唾液酸化作用也会发生改变。新近研究报道,甲状腺激素可通过降低TSH分子唾液酸化而增强其生物学活性。
1.TSH分泌的下丘脑调节:
(1)促甲状腺激素释放激素(Thyrotropin-Releasing Hormone , TRH):TRH在体内直接控制TSH的分泌过程,垂体门脉系统中的TRH在体外也可刺激TSH分泌,TRH免疫中和可导致动物模型甲减。TRH基因敲除小鼠出生后不会出现TSH的短期激增,甲状腺激素基线水平不足,而TSH对甲减反应很差。正常或甲减动物模型下丘脑室旁核破坏导致循环TRH和TSH水平降低,但低水平的TSH仍可对循环甲状腺激素水平做出适当的反应,说明TRH可决定甲状腺激素对TSH负反馈控制的调定点。
人快速静脉注射TRH后可诱发剂量依赖性的垂体TSH分泌增加,促分泌作用在注射后5min即出现,20~30min达最大程度,2h后血清TSH恢复至基线水平。更长时间(2~4h)的TRH静脉滴注可引起TSH的双时相增加,第一时相反映垂体储存的TSH的分泌,第二时相反映垂体新合成的TSH的分泌;也有学者认为血清T 3 水平增加对TSH的负反馈作用增强导致其呈现双时相分泌;也有研究证实,体外长时给予TRH可出现TSH脱敏现象,从而出现持续TRH处理时TSH水平降低的现象 [27] 。
(2)生长抑素(Somatostatin, SS):经体内外实验证实,与垂体门脉系统等浓度的SS可抑制TSH基础分泌和TRH刺激的分泌。下丘脑室旁核前区的SS浓度最高,含有SS的神经元轴突从此处伸向正中隆起;动物模型中将此处神经纤维切除,正中隆起区几乎不存在SS残留,血清TSH水平升高。同样,动物模型中免疫中和SS将使TSH水平升高,TSH对TRH反应增强。SS输注可降低人TSH脉冲分泌振幅,进行SS输注的新生儿不出现TSH产后激增峰。因此,TSH分泌受到垂体来源的TRH和SS的双向调节。
SS与垂体中特异的高亲和力的SS受体结合,SS受体有5种亚型,其中SST 2 和SST 5 存在于垂体促甲状腺细胞。SS与其受体结合后通过抑制腺苷酸环化酶而降低PKA活性,从而减少TSH分泌。SS还可通过改变细胞内Ca 2+ 浓度发挥作用。甲减时,SS抑制TSH分泌的效能下降,给予甲状腺素治疗后其抑制效能恢复。甲减小鼠模型中,SST 2 和SST 5 表达水平降低,甲状腺素治疗可诱导SST 2 、SST 5 表达。虽然短期SS输注就可以造成显著的TSH分泌抑制,但长期接受SS或其类似物治疗的患者并不会发生甲减,由此可见,SS对TSH分泌的抑制效应可能是TSH负反馈调节环的代偿机制。生长激素缺乏与TSH对TRH反应增强有关,补充生长激素可降低TRH刺激的TSH分泌,其机制可能与生长激素补充诱发下丘脑SS分泌有关。
(3)多巴胺:与垂体门脉系统等浓度的多巴胺也可抑制TSH基础分泌和TRH刺激的TSH分泌。外源性多巴胺类似物,包括不能透过血脑屏障的多巴胺类似物,可增加TSH水平。多巴胺输注降低TSH脉冲分泌振幅,使夜间TSH分泌高峰消失,但并不影响脉冲频率;给予多巴胺拮抗剂后出现相反的生物学效应。多巴胺可直接调节下丘脑激素分泌,从而间接影响TSH的分泌,有研究报道多巴胺和多巴胺受体激动剂可同时促进大鼠下丘脑TRH和SS的分泌。
多巴胺是由下丘脑弓状核神经元分泌的,分泌多巴胺的神经元轴突从弓状核延伸到正中隆起。多巴胺通过与2型多巴胺受体(DA 2 )结合作用于促甲状腺素细胞,受体激活使细胞内腺苷酸环化酶受到抑制,从而减少TSH合成和分泌。除此之外,TSH可通过诱导促甲状腺素细胞表面DA 2 的表达进行自我调控。多巴胺对TSH的抑制效应受到性激素、BMI、甲状腺功能的影响。相比于男性,多巴胺拮抗剂在女性诱发更高的TSH水平。研究表明,肥胖与TSH分泌增强有关,其机制可能是中枢多巴胺水平减低。虽然重度甲减患者TSH水平对多巴胺无反应性变化,多巴胺对TSH分泌的抑制效应在轻度甲减患者要比在正常人群中更明显。虽然短期多巴胺输注就可以造成显著的TSH分泌抑制,但长期接受多巴胺受体激动剂治疗并不会诱发甲减,这同样反映了多巴胺在TSH负反馈调节环中的代偿机制。
(4)阿片类:人体急性阿片输注可轻度升高TSH水平,吗啡拮抗剂纳洛酮(Naloxone)急性输注几乎对TSH无影响。相比而言,24h内缓慢纳洛酮滴注可使24h TSH分泌量降低,以夜间TSH脉冲振幅减低为主,TSH对TRH的反应性也降低,循环T 3 水平降低,这说明缓慢纳洛酮滴注引起的TSH水平变化足以引起甲状腺功能的变化 [28] 。以上表明,机体内源性阿片类可能是TSH的辅助刺激因子。
2.TSH分泌的外周调节:
(1)促甲状腺激素释放激素(Thyrotropin-Releasing Hormone , TRH):甲状腺激素直接阻断垂体TSH分泌,人体急性T 3 输注对TSH分泌的抑制效应可达数小时,慢性T 3 输注的抑制效应更持久。血清甲状腺激素水平在正常界值范围内的轻度变化就可以使TSH基础分泌和TRH刺激的TSH分泌发生改变,证实了垂体对甲状腺激素负反馈调节的敏感性。甲状腺激素改变TSH基础分泌水平和脉冲分泌振幅,但并不影响脉冲频率;原发性甲减患者的TSH脉冲数与健康人无差异,T 4 替代治疗可降低脉冲分泌振幅并不改变脉冲频率。
除直接抑制作用外,甲状腺激素还可通过影响下丘脑TRH、SS的分泌及垂体中相应受体的表达,从而间接地发挥抑制TSH分泌的功能。在垂体水平,甲状腺激素降低TRH受体数量,增强TRH受体降解酶的活性,从而减少TRH对TSH分泌的刺激作用。在下丘脑水平,甲减患者室旁核TRH mRNA水平升高,给予T 3 或T 4 替代治疗后TRH在室旁核的表达降低。另一方面,T 3 可直接刺激下丘脑组织分泌SS,还可增加垂体促甲状腺素细胞表面的SS受体数量。甲减大鼠模型中下丘脑SS水平降低以减少对TSH分泌的抑制作用,当给予T 3 治疗后下丘脑SS水平恢复。
(2)糖皮质激素:治疗剂量的糖皮质激素或高水平的内源性糖皮质激素抑制人和动物的TSH基础分泌水平,削弱TSH对TRH的反应,降低TSH夜间分泌高峰。健康个体输注氢化可的松使血皮质醇达到轻中度应激状态的皮质醇水平,可抑制24h TSH分泌。糖皮质激素降低TSH脉冲分泌振幅而不改变脉冲频率,可使TSH夜间分泌高峰消失。生理量的糖皮质激素也会影响TSH分泌水平,未经治疗的肾上腺皮质功能减退的患者TSH水平升高,糖皮质激素替代治疗后恢复至正常水平。动物实验证实,糖皮质激素可直接作用于垂体促甲状腺素细胞减少TSH分泌,这种直接抑制作用是糖皮质激素时间-剂量依赖性的。然而,糖皮质激素对TSH基因转录并无直接影响,接受糖皮质激素治疗的患者可维持正常的TSH脉冲频率,这提示糖皮质激素直接影响TSH分泌过程。
(3)瘦素(Leptin):瘦素主要由脂肪细胞产生,也存在于促甲状腺素细胞中,调节摄食和能量代谢,禁食状态时显著降低。外源性瘦素摄入可使大鼠TSH水平升高,机制与瘦素增加TRH基因表达、促进TRH释放有关;同样,给予禁食的人或大鼠外源性瘦素输注,可逆转禁食诱导的TSH减低。这说明,禁食引起的瘦素水平下降是引起TSH分泌抑制的重要因素。健康个体的循环TSH和瘦素存在协调的脉冲频率与昼夜节律,清晨高峰出现后,中午前出现一个最低点 [29] 。亚临床甲减患者接受左甲状腺素治疗后,血清TSH水平和瘦素水平都下降;给接受左甲状腺素抑制治疗的甲状腺癌患者注射人重组TSH可使血清瘦素水平增加。
(4)细胞因子:细胞因子是炎症反应的介导者,可由多种细胞合成并分泌,细胞因子可系统性作用于下丘脑-垂体-甲状腺轴。健康成人给予肿瘤坏死因子(Tumor Necrosis Factor, TNF)或白介素6(IL-6)输注后血清TSH水平下降,TNF和IL-1可使大鼠血清TSH水平降低。外源性细胞因子输注模拟了其他非甲状腺疾病状态下的甲状腺激素和TSH变化。TNF降低大鼠下丘脑TRH水平,抑制垂体TSH基因转录。IL-1增强大鼠大脑中的2型脱碘酶活性,通过增加垂体内T 3 减少TSH分泌。
1.TSH对甲状腺的作用:TSH通过与甲状腺细胞上的TSH受体结合而发挥其生物学作用,TSH受体是糖蛋白激素受体(Glycoprotein Hormone Receptor, GPHRs)家族的成员,GPHRs属于A型G-蛋白偶联受体亚家族。TSH受体分子结构可分为3个部分,细胞外庞大的N-端区,由3个细胞外环状结构连接的7次跨膜螺旋,细胞内短小的C-端尾。TSH分子与TSH受体结合的特异性取决于TSH β亚基、TSHβ亚基L3 β螺旋内第58~69位氨基酸序列和“安全带”区第88~105位氨基酸序列在结合并激活TSH受体及其富含亮氨酸的重复序列的过程中发挥重要作用。TSHβ亚基C-末端含有的多个赖氨酸残基(101、107、110位)和105位半胱氨酸赋予TSH分子与其受体结合的能力,TSHβ亚基半胱氨酸移码突变导致的TSH生物学活性消失可引发先天性甲减。TSHα亚基的关键序列对维持TSH的生物活性也发挥重要作用,尤其是11~20位和88~92位的氨基酸序列。α亚基天冬氨酸残基上的低聚糖链与TSH分子结合并激活受体的能力有关,α亚基天冬氨酸低聚糖链缺失的TSH分子体外生物学活性增强,但这种结构变化导致TSH体内清除增多且活性下降,类似的突变会降低CG分子与其受体的结合能力。除此之外,TSH亚基分子上的低聚糖链还参与信号传导过程。TSH受体脱敏现象与预先TSH刺激导致的cAMP水平下降有关,重组TSH受体研究表明,非甲状腺细胞表达的TSH受体不会发生脱敏,提示这种现象的产生依赖于细胞特异的辅助参与因子 [30] 。
TSH可调节甲状腺的腺体生长、细胞形态、碘代谢、甲状腺激素的生物合成等。TSHR与活化型G蛋白(GS)偶联,激活腺苷酸环化酶生成cAMP和Gq/11, cAMP和Gq/11激活PKC, PKC催化1,4,5-三磷酸肌醇及其裂解产物——磷酸肌醇的产生。TSHR分子可发生二聚化,此时TSH的结合会介导负性协同效应。TSH与TSHR同源二聚体的单体结合后,TSHR偶联GS蛋白激活cAMP;而TSH与TSHR同源二聚体的双体结合则偶联Gq/11激活PI信号通路。
TSH可通过激活JAK/STAT和mTOR/S6K1信号通路调节甲状腺细胞生长,TSH作用于甲状腺细胞的最终效应是促进以甲状腺球蛋白基因转录为起始环节的甲状腺激素的合成,虽然甲状腺球蛋白基因转录并不依赖于TSH的存在,TSH可显著提升其转录效率和甲状腺球蛋白mRNA的稳定性。TSH促进碘摄取和有机化,促进胶质腔内储存的甲状腺球蛋白发生碘化和水解,促进T 3 、T 4 的释放。
2.TSH的甲状腺外作用:患有严重的原发性甲减的青少年会出现性早熟,这提示高水平的TSH可作用于促性腺激素受体,这种交互作用已被相关研究证实,人重组TSH可以激活FSH受体,而对CG/LH受体无影响。脂肪组织也表达TSH受体,TSH可刺激体外培养的脂肪前体细胞增生并抑制其分化。眶周软组织成纤维细胞、脂肪前体细胞、脂肪细胞上TSH受体的存在或可解释TSH在Graves眼病发病机制中的作用。淋巴细胞和红细胞都有TSH及TSH受体的表达,很多学者猜测,除经典的促进甲状腺激素合成、分泌功能外,TSH还有很多其他的生物学作用。TSH激活NFκB信号通路促进IL-6释放。TSH通过调节TNF-α 和RANKL (NFκB配体的受体激活剂)的生成而抑制破骨细胞分化。大脑皮层和垂体中也存在TSH受体,星形胶质细胞和神经元有TSH受体mRNA和蛋白的表达,受体激活可促进花生四烯酸释放和2型脱碘酶活性;垂体中的TSH受体主要在星状滤泡细胞表达,可能与TSH旁分泌反馈抑制有关,也可对TSHR的自身抗体做出反应。TSH甲状腺外的生理学功能需要更多的科学研究来探讨,因为这将关系到Graves眼病新的治疗方案的安全性,也将成为甲状腺癌以TSHR为靶标的放射性同位素治疗安全性评价的科学依据。
1.血清TSH测定技术:敏感特异的血清TSH检测方法对众多甲状腺疾病的鉴别诊断和指导治疗至关重要。TSH测定方法经历了第一代放射免疫法(Radioimmunoassay, RIA)到第二代免疫分析法(Immunometric Assay, IMA)再到第三代免疫化学发光法(Immunochemiluminometric, ICMA)的演变。1926年,Uhlenhuth等发现垂体前叶中存在一种对甲状腺细胞有刺激作用的物质,直到20世纪60年代TSH得以提纯,1963年第一个抗TSH的抗体研发成功,使得放射免疫法测定血清TSH水平成为可能。放射免疫法是利用同位素标记的抗原和待测样品中未标记的抗原与抗体发生竞争性结合反应的原理进行的(图4-6),反应体系中抗体相对不足而过量的抗原被丢弃,因此放射免疫法敏感性很低,只能检测到原发性甲减患者血清中高水平的TSH。直到1982年首次报道TSH单克隆抗体技术应用于免疫学测定,这种应用两个或两个以上可识别抗原特异表位的单克隆抗体的“三明治夹心法”技术实现了TSH的精准测定。同位素、化学发光物或酶类标记的一个或多个抗体被称为“信号抗体”,另一个具有不同的表位特异性的抗体吸附在固相支持物上,被称为“捕获抗体”,测定体系中所用的抗体都是过量的,因此待测样品中TSH分子可被完全捕获,信号抗体来源的可检测信号的量与待测样品中的TSH水平直接相关。
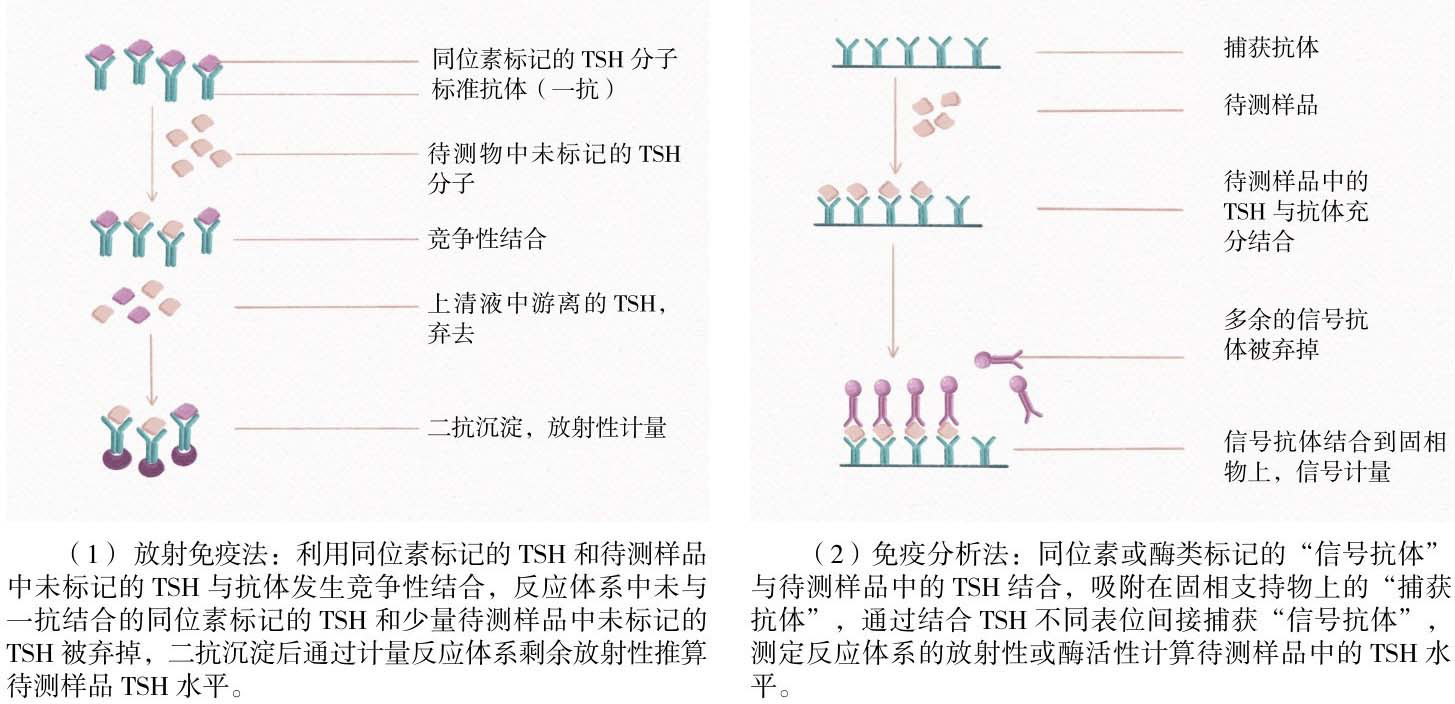
图4-6 TSH测定方法演变
TSH测定方法的改进具有重大的临床意义。首先,免疫分析法是高度特异性的,与其他糖蛋白激素不存在交叉反应;其次,免疫计量分析法可测量100%的甲状腺功能正常的对照人群的血清TSH水平,并且测定结果维持在0.45~4.1mIU/L的范围内;再者,甲亢人群与甲功正常人群的血清TSH水平几乎不存在交叉,甲亢患者极低的TSH水平和正常对照者的TSH水平可有效被鉴别。TSH检测的功能下限是指批间测定变异系数小于20%的最低TSH测定值,这个指标代表检测方法的敏感性。方法上的改进极大地降低了TSH的功能检测下限,第一代放射免疫法的功能性检测下限为0.5~1.0mIU/L,第二代测定方法降低到0.1~0.2mIU/L,第三代和第四代测定技术的功能检测下限分别为0.01~0.02mIU/L和0.001~0.002mIU/L,现今,敏感性最高的商业化测定方法是第三代。
2.血清TSH参考范围:血清TSH的正常参考范围仍存在争议。大型商业公司将来自大样本人群的测定数据的中位数±2个标准差作为TSH的正常参考范围,目前被广泛应用的TSH正常参考范围是0.5~5.0mIU/L;然而这项研究的纳入人群并未排除患有甲状腺疾病和垂体疾病的个体。美国国家健康与营养评估调查(NHANES)Ⅲ期研究进行了一项大样本人群的TSH测定研究,严格执行对象的排除标准,甲状腺疾病或垂体疾病病史、影响甲功的药物治疗史、妊娠、甲状腺自身抗体阳性等。此项研究得出的TSH正常参考范围为0.45~4.1mIU/L,平均TSH水平为1.49mIU/L,大多数正常个体的血清TSH在3.5mIU/L以下。对数据进行深入分析后发现,老年人群的TSH水平相对高于年轻人且变异范围更宽,这提示年龄增长可能与较高水平的血清TSH有关,尤其是在老年女性,然而老年人群的高TSH水平并不伴随亚临床甲减的临床症状 [31] 。一项来自澳大利亚的大型序贯研究显示,经过13年的随访,TSH水平从1.49mIU/L增加到1.81mIU/L,其中老年人群的变化最大,但TSH变化未发现性别差异。中国的一项东部地区10省市调查( n =15008)显示,血清TSH中位数为2.4mIU/L,参考值为0.76~6.92mIU/L,显著高于美国国家健康与营养评估调查(NHANES)的报告。分析其原因可能与碘摄入量增加有关(MUI196μg/L vs 145μg/L)。
虽然人群的TSH正常参考范围相对较宽,个体的TSH变化程度很小,血清TSH水平在多个因素的精密调控下围绕个体调定点发生微小波动。一项研究选取了15例甲状腺功能正常的女性,每个月进行TSH水平测定共持续1年,结果显示,不同个体的TSH调定点相差很大,但个体的TSH水平围绕自身调定点水平聚集分布。TSH调定点的决定因素尚不清楚,有单卵孪生子研究提示TSH调定点主要由遗传背景决定。基因分析研究揭示了多个基因与TSH调定点有关,但并未发现发挥主要调节作用的单个基因。
影响甲功正常的健康人群血清TSH的主要环境因素是碘营养水平,尿碘增加是血清TSH水平升高的相关因素。滕卫平课题组报道的中国流行病学数据显示,碘超足量地区亚临床甲减(TSH>4.2 mIU/L)发病率较碘足量地区显著升高(22.6% vs 12.7%) [32] 。该研究组的另一项研究在轻度碘缺乏(盘山)、碘超足量(彰武)、碘过量(黄骅)地区选取的大样本人群( n =3.761)数据显示,随着碘摄入量的增加,血清TSH水平显著增加 [33] ,这提示TSH正常参考范围的确定应考虑到各地碘营养水平的差异性。高碘可能通过抑制下丘脑内2型脱碘酶的活性从而减少T 4 转换为T 3 ,进而减少对下丘脑内TRH分泌细胞的负反馈抑制,从而使血清TSH水平升高 [34] ;又有学者报道,高碘摄入并不降低下丘脑内2型脱碘酶的mRNA水平,而是通过改变与2型脱碘酶相关的泛素化酶活性,而增加下丘脑水平2型脱碘酶蛋白降解 [35] 。
3.TSH游离α亚基和β亚基的测定:1974年,TSHα亚单位和β亚单位从人TSH中提纯出来,继而针对不同亚单位的特异性抗体成功开发。继放射免疫法之后,免疫学测定方法成为游离α亚单位的主要测定方式。而游离的TSHβ亚单位一般只在原发性甲减的患者中可检测到,因此测定游离β亚单位的临床价值有限。游离α亚单位水平可作为诊断垂体和胎盘疾病的参考指标,因为即使在甲状腺功能正常和性腺功能正常的群体,血清游离α亚单位水平也是可测量的。患有以下疾病的患者呈现出较高的血清游离α亚基水平:促甲状腺激素或促性腺激素分泌功能的垂体瘤、绒毛膜癌、肺癌、胰腺癌、胃癌、前列腺癌、卵巢癌等。原发性甲减和原发性性腺功能不足与较高的游离α亚单位水平有关,分别反映了血清中高水平TSH和高水平促性腺激素的存在。
4.TRH兴奋实验:TRH兴奋实验是临床上常用的甲状腺疾病的鉴别诊断方法。静脉给予200~500μg TRH,分别于注射后0min、30min、60min、120min、180min时间点采血测定血清TSH水平,可以反映垂体对TRH的反应能力,用于评估下丘脑-垂体-甲状腺轴的调节功能。甲状腺功能正常的个体,血清TSH注射后20~30min达到峰值,峰值为基础值的5~10倍。原发性甲亢的患者由于高水平的甲状腺激素对TSH的抑制作用,表现为TSH对TRH的无反应。相反,原发性甲减的患者表现为对TRH的强反应,TSH水平显著升高。垂体疾病或下丘脑疾病所致的中枢性甲减患者,也表现为TSH对TRH的无反应。TSH分泌性垂体瘤患者接受TRH刺激后,TSH水平略升高,小于基础水平的2倍。
对老年人群的血清TSH水平变化目前仍存在争议,有研究显示年龄增加与高TSH水平有关,而也有研究得出相反的结论。这种分歧很大程度上取决于研究人群的异质性,TSH水平在正常参考范围之外的个体是否排除、所研究地区的碘营养水平是否一致都会影响研究结论。尽管争议存在,TSH水平确实会随年龄变化而改变,因此很多学者致力于更科学、精确的TSH参考范围的制定。来源于不同年龄段、不同种族、不同性别的大样本人群的测定结果经系数加权后得出的TSH分布曲线是目前计算TSH正常参考范围的基础。美国国家健康与营养评估调查(NHANES)Ⅲ期纳入17353个研究对象、持续6年(1988—1994)的研究为TSH正常参考范围的确立提供了重要的数据。此项研究得出的TSH分布图第97.5百分位数是4.12mIU/L,由此4.5mIU/L被人为规定为TSH参考范围上限 [31] 。年龄大于70岁的人群分组中,TSH高于参考范围上限的发生率增高,最高可达15%;在美国,年龄大于70岁的个体有35000000,也就是说,按照这个给定的参考范围,将有5250000人被诊断为亚临床甲减。不同年龄、性别、种族人群的中位TSH虽然差异较小,但达到统计学差异,因此建议针对不同人群亚组建立特异性的TSH。
血清TSH水平随年龄增长而升高的现象是建立于传统的TSH分布曲线和据此推算出的参考范围上限基础上的,而这个分布曲线并不是针对特定年龄亚组的人群,而是来源于不同年龄段和不同种族的大样本人群。2002年,美国国家临床检验学会发布TSH测定方法和标准的指南共识,也包括TSH正常参考范围确定的方法;指南推荐:TSH正常参考范围应根据至少包括120名无甲状腺疾病相关病史的个体的参考人群TSH浓度而制定,这些个体应排除甲状腺肿、甲状腺疾病既往史或家族史、影响甲状腺功能的用药史或血清抗体阳性等情况。然而,根据指南推荐的方法得出的TSH分布曲线并不呈高斯分布,而是向高TSH浓度方向偏移,即使经过对数转换后也是如此 [36] 。
对TSH分布曲线偏移现象的一种解释是,目前广泛应用的TSH分布曲线是美国国家临床生化学会(National Academy of Clinical Biochemistry, NACB)指南根据美国全体人群制定的,实际上就是各种异质性人群TSH分布曲线的综合。因此,针对不同年龄、不同种族人群亚组的特异性TSH分布曲线就会与这个全人群的综合曲线产生偏移。那么,基于这个综合后的TSH分布曲线推算出的TSH参考范围若应用于特定年龄、种族的人群亚族,将会使TSH水平原本在其人群特定的正常参考范围之内的个体被诊断为TSH升高。例如,NHANESⅢ期研究报道,不患有甲状腺疾病的人群的血清TSH水平随年龄增长有显著增高的趋势,剔除掉血清甲状腺自身抗体阳性的个体后仍然得到相同的结论。因为TSH正常参考上限是基于综合后的TSH分布曲线推算得出,因此血清TSH水平高于这个上限的14%的个体被认为患有甲减或亚临床甲减。但是,如果根据老年人群特异的TSH分布曲线(向较高TSH水平方向偏移的分布曲线)推算出其特异的参考范围去诊断,其真实的TSH水平将低于其所属人群亚族特异的参考上限,因而不会被诊断为甲减或亚临床甲减。对NHANESⅢ期和NHANES 1999—2002年的调查数据进行分析,结果显示,TSH分布曲线和参考值范围在各年龄段人群都是特异的,并随年龄增加向TSH较高水平方向偏移。若应用年龄特异的TSH参考范围诊断,约50%被传统TSH参考范围定义为甲减或亚临床甲减的不患有甲状腺疾病。同样,随年龄增长TSH分布曲线向高TSH浓度方向偏移的现象在黑种人、高加索人、西班牙人中也有报道。
血清TSH水平除随年龄变化外,也会有种族差异,例如,黑种人的中位TSH水平低于高加索人。与白种人相比,黑种人和西班牙人的TSH分布曲线向低TSH浓度方向偏移。由于TSH有年龄和种族差异,因此人们呼吁建立人群特异性的TSH分布曲线和参考范围。应用年龄和种族特异性的TSH参考范围,将减少轻度TSH异常的误诊率。
2011年,我国一项纳入2488人的流行病学调查得出的总人群的TSH参考范围为0.48~5.50mIU/L [ISH均值:(1.58±1.92)mIU/L;年龄:40.37±15.88]。将入组研究对象按年龄分组,发现年龄小于20岁的人群亚组血清TSH水平显著高于其他年龄亚组,而其他各个年龄分组组间比较都未见明显差异。作者将年龄小于20岁的个体排除后得到的TSH正常参考范围为(0.46~5.19)mIU/L[ n =2.118, TSH均值:(1.49±1.91)mIU/L] [37] 。
重组人TSH(Recombinate Human TSH, rhTSH)是通过胚胎肾细胞或中国仓鼠卵巢CHO细胞系内稳定转染TSHα亚单位和β亚单位基因而产生。这些细胞不能表达垂体特异的N-乙酰半乳糖胺转移酶和GalNAc-磺基转移酶,因此相比于垂体提取的天然TSH, rhTSH唾液酸化修饰水平较高,不含N-乙酰半乳糖胺以及硫酸盐末端,而是构成了以唾液酸为末端的寡糖链。两次体外生物活性检测显示,rhTSH的最大刺激活性与天然TSH类似,但根据半效应剂量评估的生物学效能略低于天然TSH。rhTSH在大鼠体内的清除率降低,仅为天然人TSH清除率的一半。rhTSH的在体半衰期远远长于天然TSH,末次输注48h后仍可检测到持续的高水平的rhTSH,而天然TSH的半衰期为1~1.5h。rhTSH的低效能、低清除率、长半衰期等生物学活性特点与其分子内唾液酸含量的增加有关 [38] 。
随着对TSH分子结构的进一步了解和医学技术的进步,rhTSH的规模化生产已经实现。相比于解剖手段从垂体中提取和纯化的天然TSH, rhTSH同源性好、纯度更高,不存在垂体其他糖蛋白激素或相应裂解物的污染。rhTSH的异质性主要源于其6个分子异构体的存在,它们因糖链内唾液酸残基末端的数目不同而形成。rhTSH成功应用于临床并体现出良好的临床价值,尤其在复发性甲状腺癌的诊断和放射性碘治疗的前期准备方面有很好的前景。