
下载掌阅APP,畅读海量书库
立即打开



有机化学是用分子结构来描述的一门学科,结构决定性质,性质决定用途。因此,有机化合物的结构鉴定是有机化学研究工作中一项最基础和最重要的工作。早期的有机化合物结构鉴定,主要是通过化学方法实现的,对于比较复杂的分子来说,需要通过多种化学反应,分析大量的样品,实验工作比较烦琐,往往需要较长的时间才能完成。测定有机化合物结构的波谱法,是20世纪五六十年代发展起来的现代物理实验方法,具有快速、准确、样品用量少等优点,目前已经成为化学研究工作中不可缺少的研究手段。
有机化合物结构鉴定常用的波谱主要有紫外光谱(ultraviolet spectrum,UV)、红外光谱(infrared spectrum,IR)、核磁共振谱(nuclearmagnetic resonance,NMR)和质谱(mass spectrum,MS)。
紫外光的波长为10~400nm。分子吸收紫外光后,发生价电子能级跃迁,产生紫外吸收光谱,因此,紫外光谱又称电子光谱。远紫外光波谱测定比较困难(易被空气中氧气和二氧化碳吸收),通常所说的紫外光谱是指近紫外区(200~400nm)的吸收光谱。有些有机分子特别是具有共轭体系的分子的价电子跃迁吸收往往出现在可见光区(400~800nm)。从应用的角度考虑,通常将紫外和可见光谱连在一起,称为紫外-可见光谱。紫外光谱的产生机理可参阅相关教材。
现在紫外光谱图(见图1-7)一般都是由仪器检测终端相连的计算机自动生成的。紫外光谱图提供了两个重要的数据:吸收峰的位置和吸收光谱的吸收强度。在大多数的文献报道中,通常不绘制紫外光谱图,只是报道化合物的最大吸收峰的波长λ max 及与之相对应的摩尔消光系数ε max 。由于用于紫外光谱测定的试样的溶剂可能影响λ max 和ε max 值,文献报道化合物的紫外光谱图及λ max 、ε max 时,需标明所用的溶剂。
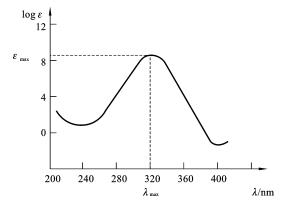
图1-7 紫外光谱图
紫外光谱可以提供分子中生色基团和助色基团的信息。不共轭的生色基团的紫外吸收波长大多在远紫外区,当分子中存在共轭(p-π共轭和π-π共轭)结构时,紫外吸收波长落在近紫外区。共轭双键越多,吸收波长越长,共轭双键增加到一定程度,吸收波长可进入可见区。因此,紫外光谱是判断分子内是否含有共轭结构的最有效的手段。
1.紫外-可见分光光度计
紫外光谱是用紫外-可见分光光度计测定的。紫外-可见分光光度计有单光束、双光束和双波长等几种类型。目前使用的大多为双光束型,其光路图如图1-8所示。
紫外-可见分光光度计由下列部件组成:
(1)光源 在整个紫外光区或可见光谱区可以发射连续光谱,具有足够的辐射强度、较好的稳定性、较长的使用寿命。可见光区通常用钨灯作为光源,其辐射波长范围为320~2500nm。紫外光源通常用氘灯,能发射185~400nm的连续光谱。
(2)单色器 将光源发射的复合光分解成单色光,并可从中选出一任意波长单色光的光学系统。主件为棱镜或光栅。
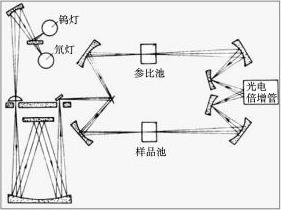
图1-8 双光束紫外-可见分光光度计光路图
(3)样品室 由各种类型的吸收池(比色皿)和相应的池架附件构成。吸收池主要有石英池和玻璃池两种。紫外区必须采用石英池,可见光区可用玻璃池。
(4)检测器 利用光电效应将透过吸收池的光信号变成可测的电信号的装置,常用的有光电池、光电管或光电倍增管。
(5)结果显示记录系统 目前一般都是用计算机进行仪器自动控制和结果处理。
2.紫外光谱的样品测试
紫外光谱的样品测试一般按下列步骤进行:
(1)选择溶剂 紫外光谱测定常用的溶剂有烷烃(己烷、庚烷、环己烷)、H 2 O、甲醇、乙醇等。测定非极性化合物多用烷烃作溶剂,而测定极性化合物多用H 2 O、甲醇、乙醇作溶剂。
溶剂需要选用光谱纯的,若没有光谱纯的溶剂,可用普通溶剂除去相关杂质后使用。如烷烃溶剂中往往含有烯烃或芳烃杂质,可用硅胶吸附法除去,或加入适量浓硫酸振摇洗涤后放置24h,分去硫酸,再依次用NaOH和水洗涤,再用CaCl 2 干燥后蒸馏备用。乙醇中可能存在醛类杂质,可加入10%固体NaOH和少量AgNO 3 ,放置4h,然后回流加热1h后蒸馏除去。氯仿中的稳定剂(乙醇)或光气可用浓硫酸洗涤除去。水作溶剂时,要用新鲜的蒸馏水或去离子水,且不要贮存在塑料瓶中。
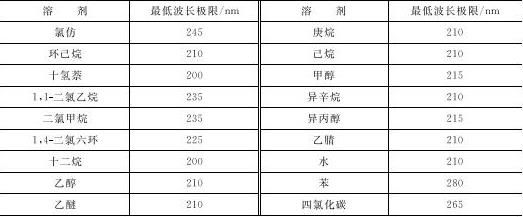
选择溶剂还要求溶剂必须在测量波段是透明的,否则会发生吸收造成干扰。表1-2列举了常用溶剂的使用波长极限,在极限以上溶剂是透明的,在极限以下则有吸收,会发生干扰。
表1-2 常用溶剂的最低使用波长极限
(2)溶液的配制 一般溶液的浓度最好使透射比为20%~65%,以10 -5 ~10 -2 mol/L为宜。有π→π*跃迁的样品的摩尔吸收系数ε很大,因此样品的浓度必须很低,一般是10-5~10-4mol/L。如果酸性或碱性物质用水作溶剂,由于其离解的阴离子或阳离子的光谱与母体不同,会出现混合光谱,因此酸性物质应在0.1mol/LhCl水溶液中进行,碱性物质则在0.1mol/L NaOH水溶液中进行。
(3)测定 操作时在样品池内装满样品溶液,将盖子沿池口边缘轻轻平推盖好,石英池表面的溢出物要用软的薄绢或擦镜纸揩拭,手指切不可触及石英池的光学表面。样品池放入样品室后,按仪器说明书提供的操作步骤进行测试。
1.吸收带
分子吸收光能使电子发生能级的跃迁时,伴随着振动能级和转动能级的变化,因此,紫外光谱图由吸收带组成。紫外光谱图中常见的吸收带有R吸收带、K吸收带、B吸收带、E吸收带等几种。
R吸收带为n→π*跃迁所引起的吸收带。如>C=O、—NO 2 、—CHO等,其特点为吸收强度弱。ε max 不超过200,一般在100以内,吸收峰波长一般在270nm以上。
K吸收带为π→π*跃迁所引起的吸收带,由共轭双键产生。该带的特点为吸收峰很强,ε max >10000(lgε>4)。共轭双键增加,λmax向长波方向移动,ε max 随之增加。在同一化合物中K吸收带的吸收波长比R吸收带的短。
B吸收带为芳环的π→π*跃迁所引起的吸收带,为一宽峰,其波长为230~270nm,中心在254nm,ε max 约为204。该吸收带常因与芳环振动吸收的叠加而分裂为多重小峰的精细结构,该结构可用于识别芳香族化合物。
E吸收带是芳香族化合物的另外一个特征吸收带,也为π→π*跃迁。E吸收带又分为E 1 吸收带和E 2 吸收带。E 1 吸收带波长约为184nm,ε max 约为60000;E 2 吸收带波长为204nm,ε max 约为7900。E 1 吸收带、E 2 吸收带都属于强吸收。
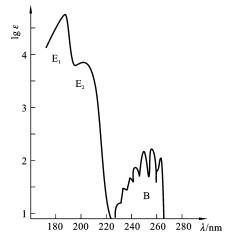
图1-9 苯在异辛烷溶剂中的紫外吸收光谱
图1-9所示的为苯在异辛烷溶剂中的紫外吸收光谱。
2.结构测定
通过测定样品化合物特性参数λmax和ε max ,然后与文献数据进行比较,可以作为结构测定依据之一。根据紫外光谱还可了解样品分子的共轭程度、空间效应、氢键等;也可对饱和与不饱和化合物、异构体及构象进行判别。
(1)若在200~750nm波长范围内无吸收峰,则可能是直链烷烃、环烷烃、饱和脂肪族化合物或仅含一个双键的烯烃等。
(2)若在270~350nm波长范围内有低强度吸收峰(ε=10~100L/mol·cm)(n→π跃迁),则可能含有一个简单非共轭且含有n电子的生色基团,如羰基。
(3)若在250~300nm波长范围内有中等强度的吸收峰,则可能含苯环。
(4)若在210~250nm波长范围内有强吸收峰,则可能含有2个相互共轭的双键;若在260~300nm波长范围内有强吸收峰,则说明该有机物可能含有3个或3个以上相互共轭的双键。
(5)若该有机物的吸收峰延伸至可见光区,则该有机物可能是长链共轭或稠环化合物。
(6)与标准物质的吸收光谱进行比较。目前《The sadtler standard spectra(Ultraviolet)》标准谱图库已收集了46000多种化合物紫外光谱的标准谱图,在相同的实验条件(仪器条件、溶剂)下,将未知物的紫外光谱与标准物质的紫外光谱进行比较。若两者谱图相同,则可认为含有相同的生色基团,但要注意不一定是相同的物质,紫外光谱只是确定结构的辅助工具。
3.物质纯度的检查
用紫外光谱法测定物质纯度有其独特的优点。因为含共轭体系的化合物有很高的紫外检测灵敏度,而饱和或某些含孤立双键的化合物则没有紫外吸收,利用这种选择性,在下列两种情况下紫外光谱可方便地检查物质纯度:一是样品化合物在近紫外区一定波长范围内没有吸收,而杂质在该波长范围有特征吸收;二是样品化合物在近紫外或可见光区有吸收,而杂质没有吸收,这样就可通过比较等浓度的纯度待定样品和其纯物质样品的吸收强度来确定纯度待定样品的纯度。
红外光谱是由化合物分子吸收红外光时,振动能级和转动能级发生跃迁而产生的吸收光谱,属于分子光谱。研究有机化合物分子结构的红外光谱处于中红外和近红外区,波数位于400~4000cm-1(波长为2.5~25μm)。
红外光谱的产生机理可参阅相关教材。一张红外光谱图,通常呈现多个吸收峰,各个峰的强弱不同,形状各异(宽峰、尖峰、肩峰、双峰等)。根据吸收峰的波长或波数的位置及峰的形状,可以获得有关有机化合物分子官能团或分子结构的信息。
1.红外分光光度计(红外光谱仪)
红外光谱是由红外分光光度计测定的。目前使用的红外分光光度计主要有两种类型:一种是色散型;另一种是干涉型(傅里叶变换)。
色散型红外分光光度计的结构和紫外-可见分光光度计大体一样,也由光源、吸收池、单色器、检测器以及记录显示装置组成。红外分光光度计的样品池是放在光源和单色器之间,而紫外-可见分光光度计的样品池则是放在单色器的后面。
(1)光源 红外光谱仪中所用的光源通常是一种惰性固体,用电加热使之发射高强度的连续红外辐射。常用的是能斯特灯或硅碳棒。能斯特灯是用氧化锆、氧化钇和氧化钍烧结而成的中空棒和实心棒。工作温度约为1700℃,在此高温下导电并发射红外线,但在室温下是非导体,因此,在工作之前要预热。它的特点是发射强度高,使用寿命长,稳定性较好。缺点是价格比硅碳棒贵,机械强度差,操作不如硅碳棒方便。硅碳棒是由碳化硅烧结而成,工作温度为1200~1500℃,在低波数区发光强度大,坚固。
(2)样品池 因玻璃、石英等材料不能透过红外光,红外光谱仪样品池要用可透过红外光的NaCl、KBr、CsI、KRS-5(Tl 58%、TlBr42%)等材料制成窗片。最常用的是用NaCl单晶材料制成的窗片,因其容易吸水溶解,所以应注意防潮。
(3)单色器 由色散元件、准直镜和狭缝构成。色散元件常用复制的闪耀光栅。由于闪耀光栅存在次级光谱的干扰,所以通常是将光栅和用来分离次光谱的滤光器或前置棱镜结合起来使用。
(4)检测器 常用的红外检测器有高真空热电偶、热释电检测器和碲镉汞检测器。前二者运用热电效应的原理,后者运用光电效应的原理。
(5)结果显示记录系统 目前一般都是用计算机进行仪器自动控制和结果处理。
傅里叶变换红外光谱仪(FTIR)没有色散元件,主要由光源(硅碳棒、高压汞灯)、迈克尔逊干涉仪、检测器、计算机和记录仪组成。核心部分为迈克尔逊干涉仪,它将光源来的信号以干涉图的形式送往计算机进行傅里叶变换,最后将干涉图还原成光谱图。它与色散型红外光度计的主要区别在于干涉仪和电子计算机两部分。
傅里叶变换红外光谱仪具有以下优点,正在被越来越广泛地使用。
(1)扫描速度快(几十次/秒),信号累加,信噪比高。
(2)光通量大,所有频率同时测量,检测灵敏度高,样品用量少。
(3)扫描速度快,可跟踪反应历程,作反应动力学研究,并可与气相色谱、液相色谱联用。
(4)测量频率范围宽,可达到4500~6cm -1 ,杂散光少,波数精度高,分辨率可达0.05cm -1 。
(5)对温度、湿度要求不高。
(6)光学部件简单,只有一个动镜在实验中运动,不易磨损。
2.红外光谱的样品测试
红外光谱的试样可以是液体、固体或气体,一般应符合下列要求:
(1)试样应该是单一组分的纯物质,纯度应大于98%或符合商业规格,这样才便于与纯物质的标准光谱进行对照。多组分试样应在测定前尽量预先用分馏、萃取、重结晶或色谱法进行分离提纯,否则各组分光谱相互重叠,难于判断。
(2)试样中不应含有游离水。因为水本身有红外吸收,会严重干扰样品的图谱,同时还会侵蚀样品池的盐窗。
(3)试样的浓度和测试厚度应选择适当,以使光谱图中的大多数吸收峰的透射比处于10%~80%。气态样品可在玻璃气槽内进行测定。玻璃气槽为一密封的容器,两端粘有红外透光的NaCl或KBr窗片。引入样品前,将气槽抽真空,再将试样注入。
液体样品可在液体样品池内测定。样品池的两侧是用NaCl或KBr等晶片做成的窗片。
沸点较高、不易清洗的液体样品可采用液膜法测定,该法是定性分析中常用的简便方法。具体操作是在可拆样品池的两片盐窗片之间,滴上1~2滴液体样品,形成一薄膜。液膜厚度可借助于样品池架上的固紧螺丝作微小调节。但是低沸点易挥发的样品不宜采用此法。
固体样品通常可采用下列方法制样:
(1)压片法将1~2mg试样与200mg纯KBr研细均匀,置于模具中,用(5~10)×10 7 Pa压力在压片机上压成透明薄片,即可用于测定。试样和KBr都应经干燥处理,研磨到粒度小于2μm,以免散射光影响。
(2)石蜡糊法将干燥处理后的试样研细,与液状石蜡或全氟代烃混合,调成糊状,夹在盐窗片中测定。
(3)薄膜法主要用于高分子化合物的测定。可将它们直接加热熔融后涂制或压制成膜;也可将试样溶解在低沸点的易挥发溶剂中,涂在盐窗片上,待溶剂挥发后成膜测定。
红外光谱在结构测定方面的应用,可分为两种情况:一种是已知化合物的鉴定;另一种是未知化合物的鉴定。
已知化合物的鉴定比较容易,就是将所测得的样品的红外光谱(见图1-10)与标准图谱库或文献报道的该化合物的图谱进行对比。若两张谱图完全相同,就可以确定该化合物与标准图谱或报道的化合物是同一种物质;若两张谱图不一样,或峰位不一致,则说明两者可能不为同一化合物,或样品有杂质。使用文献上的谱图应当注意试样的物态、结晶状态、溶剂、测定条件以及所用仪器类型均应与标准谱图或报道的图谱相同。
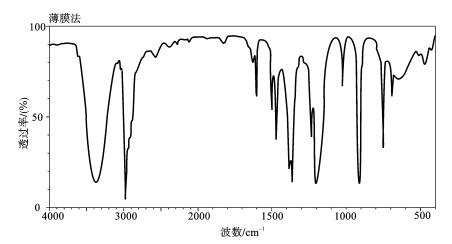
图1-10 叔丁醇的红外光谱图
目前可检索的标准图谱库有萨德勒(Sadtler)标准红外光谱图、Aldrich试剂公司的Aldrich红外谱图库、Sigma试剂公司的Sigma Fourier红外光谱图库、分子光谱文献“DMS”(documentation ofmolecular spectroscopy)穿孔卡片、美国石油研究所(API)编制的“API”红外光谱资料等。
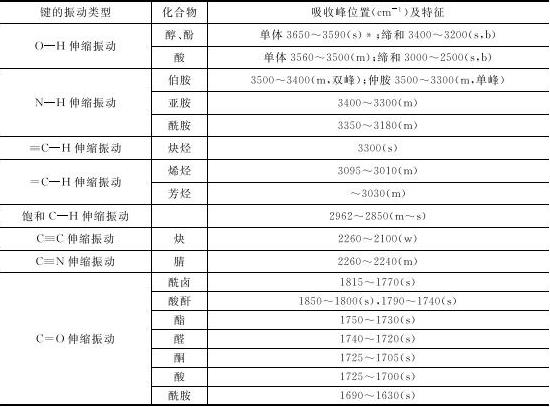
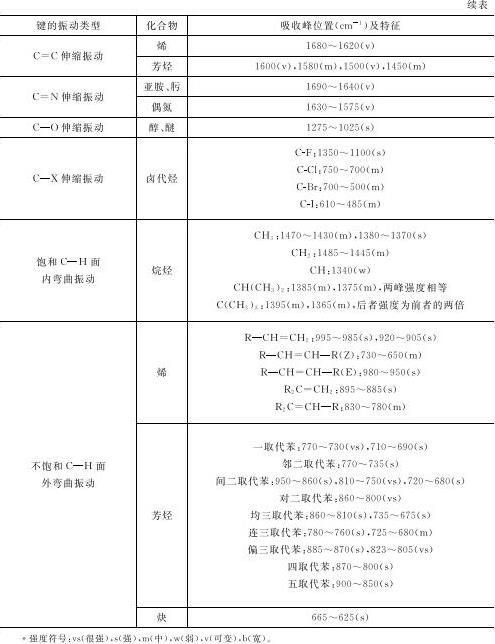
未知化合物的情况比较复杂,需要通过解析所测得的红外光谱图来判断所测试样品分子中有哪些基团(官能团)存在,判断基团是否存在主要是利用这些基团在红外光谱图中的特征吸收位置(见表1-3)。有时也可从红外光谱图获得这些基团在分子中的相对位置的信息。实际上,单用红外光谱图很难确定一个未知化合物的结构,往往需要结合其他测试手段才能实现。
表1-3 常见官能团的红外特征吸
核磁共振谱是利用磁矩不为零的原子核,在外磁场作用下自旋能级发生分裂,共振吸收某一定频率的电磁波射频辐射(处于无线电波的振动频率区间)所得到的图谱。核磁共振技术是有机化合物结构鉴定的重要手段,测定结果能为确定官能团和烃基在分子中的排列情况提供十分有用的信息。常用的核磁共振谱有氢谱和碳谱,氢谱常用英文缩写 1 HNMR或PNMR(质子核磁共振英文缩写)表示,碳谱常用 13 CNMR表示。由于 1 HNMR谱是最常用的,这里只讨论 1 HNMR谱。
核磁共振谱的产生机理可参阅相关教材。在核磁共振测量中,以吸收能量的强度为纵坐标,化学位移δ为横坐标绘出一系列吸收峰,由此得到的波谱图就是核磁共振谱图,如图1-11所示。
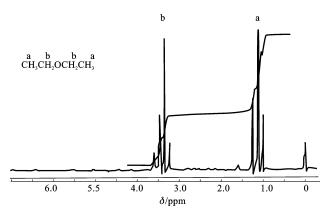
图1-11 乙醚的高分辨率核磁共振谱图
1.核磁共振仪
核磁共振仪按扫描方式不同,可分为两大类:一类是连续波核磁共振仪,其射频的频率或外磁场的强度是连续变化的,即进行连续扫描一直到被观测的核依次被激发发生核磁共振;另一类是脉冲傅里叶变换核磁共振仪,这类仪器采用恒定的磁场,用一定频率宽度的射频强脉冲辐照试样,激发全部欲观测的核,得到全部共振信号。当脉冲发射时,试样中每种核都对脉冲单个频率产生吸收。
核磁共振仪主要由磁铁、射频发生器、射频接收器、记录仪等组成,如图1-12所示。
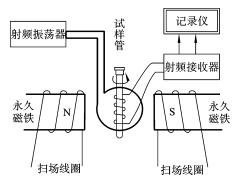
图1-12 核磁共振仪原理示意图
(1)磁铁磁铁或磁体产生强的静磁场,以满足产生核磁共振的要求。100mHz以下的低频谱仪采用电磁铁或永久磁铁。200mHz以上高频谱仪采用超导磁体,它利用了含铌合金在液氦温度下的超导性质。由含铌合金丝缠绕的超导线圈完全浸泡在液氦中间。为了降低液氦的消耗,其外围是液氮层。液氦及液氮均由高真空的罐体贮存,以降低蒸发量。液氦需及时补充,视不同谱仪而定,为3至10个月。每7至10天需补加液氮。因此,超导核磁共振仪的价格及日常保养费用都很高。扫场时,磁场强度的变化通过调节扫场线圈中的电流来实现。
(2)射频振荡器(射频发生器)用于产生射频电磁波照射样品。
(3)射频接收器射频接收器线圈在试样管的周围,并与振荡器线圈和扫描线圈相垂直。当射频振荡器发生的频率v 0 与磁场强度H 0 达到前述特定组合v 0 =γH 0 /(2π)时,放置在磁场和射频线圈中间的试样就要发生共振而吸收能量。这个能量的吸收情况通过射频接收器检出,经过放大后记录下来。所以核磁共振波谱仪测量的是共振吸收。
(4)记录仪目前核磁共振仪的记录装置都是用计算机进行仪器自动控制和结果处理的。计算机接收射频接收器发出的经过放大后的检测信号,经过数据处理得到核磁共振谱,还能自动画出积分线,给出各组共振吸收峰的相对面积。
2.核磁共振的样品测试
核磁共振实验时样品管放在磁极中心,磁铁应该对样品提供强而均匀的磁场。但实际上磁铁的磁场不可能很均匀,因此需要使样品管以一定速度旋转,以克服磁场不均匀所引起的信号峰加宽。
做质子核磁共振谱( 1 HNMR谱)时,常用外径为6mm的薄壁玻璃管。测定时样品常常被配成溶液,这是由于液态样品可以得到分辨率较好的图谱。要求选择不产生干扰信号、溶解性能好、稳定的氘代溶剂。溶液的浓度应为2%~10%。样品的溶液应有较低的黏度,否则会降低谱峰的分辨率。如纯液体黏度大,应用适当溶剂稀释或升温测谱。常用的溶剂有CCl 4 、CDCl 3 、(CD 3 ) 2 SO、(CD 3 ) 2 CO、C 6 D 6 、D 2 O等。
为测定化学位移值,需加入一定的基准物质。基准物质加在样品溶液中称为内标。若出于溶解度或化学反应性等的考虑,基准物质不能加在样品溶液中,可将液态基准物质(或固态基准物质的溶液)封入毛细管再插到样品管中,称为外标。
基准物质最常用的是四甲基硅烷(TMS)。TMS的浓度一般为0.2%左右。
根据有机化合物的核磁共振谱图,可以得到化合物结构的信息:
(1)由信号峰的组数可以推知有机化合物分子中有几类氢;
(2)由信号峰的强度(峰面积或积分曲线高度),可以知道化合物中各类氢的数目的相对比,再根据分子中氢的总个数判断各类氢原子的数目;
(3)从各信号峰的裂分数目可以推知其相邻氢的数目;
(4)由各峰的化学位移(见表1-4)可以推知各类型氢的归属。
表1-4 特征质子的化学位移
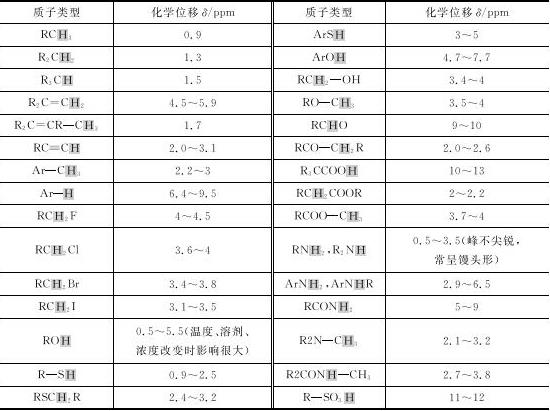
(5)由裂分峰的外形或偶合常数(相邻两个小裂分峰之间的距离称为偶合常数,用J表示,单位为Hz),可以判断哪种类型H所连的碳原子是相邻的,因为相互偶合的氢原子吸收裂分峰的偶合常数相等。
核磁共振谱在结构测定方面的应用,也分为两种情况:一种是已知化合物的鉴定;另一种是未知化合物的鉴定。
已知化合物的鉴定比较容易,就是将所测得样品的核磁共振谱与标准图谱库或文献报道的该化合物的图谱进行对比。若两张谱图完全相同,就可以确定该化合物与标准图谱或报道的化合物是同一种物质;若两张谱图不一样,或峰位不一致,则说明两者可能不为同一化合物,或样品有杂质。使用文献上的谱图应当注意测试时所用的溶剂、测定条件以及所用仪器类型均应与标准谱图或报道的图谱相同。目前可检索的标准图谱库有萨德勒(Sadtler)标准核磁共振谱图等。
未知化合物的情况比较复杂,需要通过解析所测得的核磁共振谱图来判断所测试样品分子中有哪类官能团的氢原子存在,也可根据吸收峰的裂分情况和偶合常数等获得这些基团在分子中的相对位置的信息。实践中,单用核磁共振谱图确定一个未知化合物的结构比较困难,往往需要结合其他测试手段才能实现。
质谱分析法是通过对被测样品离子的质荷比(m/Z)的测定来进行分析的一种分析方法。被分析的样品通过仪器进行离子化后,利用不同离子在电场或磁场的运动行为的不同,把离子按质荷比分开而得到质谱。通过对样品的质谱进行分析,可以得到样品分子结构的有关信息。
质谱分析具有快速、简捷、精确,样品用量少等优点。通过色谱和质谱联用,还可以测定混合物的组成及各组分的相对分子量,并获得与推导结构有关的信息。
质谱是样品的不同质量的分子碎片的质量分布图,其原理可以借助于质谱仪的工作原理图(见图1-13)予以说明。
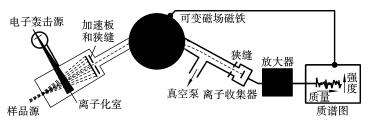
图1-13 质谱仪的工作原理图
汽化的样品进入高真空的离子化室,用具有一定能量的电子束(电子轰击源、EI源)轰击气态分子,分子失去电子成为带正电荷的分子离子M + ,获得电子束能量的激发态分子离子会继续发生化学键的断裂,生成阳离子、阴离子或不带电的碎片。产生带正电荷的离子经电位差为几百到几千伏的电场加速,进入分析系统。分析系统的可变磁场对正离子产生洛伦茨力作用,使每一种离子按照一定的轨道继续前进,其行进轨道的曲率半径取决于各种离子的质荷比。这样,分子离子、碎片可按质荷比的大小得到分离。所有相同质荷比的离子结合在一起,形成离子流,各种离子流沿不同的曲率半径轨道通过狭缝进入离子收集、检定系统。各种离子流出现不同信号,其强度与离子流成正比,记录所产生的信号,即得到样品的质谱。通常正离子的电荷为1,故所记录的信号即是不同质量的正离子。M + 的质量就是化合物的相对分子质量;将碎片的种类、质量和强度与化合物键合规律结合起来,就可以推断分子的结构。
分子被电子束轰击时的裂解规律可参阅相关教材。
1.质谱仪
目前用于质谱测量的质谱仪有多种类型,如磁质谱仪、四极质谱仪、离子阱质谱仪、飞行时间质谱仪和离子回旋共振傅里叶质谱仪等。各种质谱仪具有不同的特点和适用范围,一般都由下面几个系统组成。
(1)真空系统 质谱仪的离子源、质谱分析器及检测器必须处于高真空状态(离子源的真空度应达10 -5 ~10 -3 Pa,质量分析器应达10 -6 Pa),通常用机械泵预抽真空,然后用扩散泵高效率并连续地抽气。
(2)进样系统 进样系统一般可分为下列三种情况。
①直接进样(静态法):对气体或挥发性液体的纯化合物,可用微量注射器注入直接进样到离子化室。
②探针进样:对于挥发性很小的固体样品,需将样品放在不锈钢杆或探针顶端的小杯内,将探针通过样品加入口放进离子化室中,然后加热离子化室直至固体挥发。
③色谱进样:对于一些组分较复杂的混合物,需将样品分离成一个个单一组分,再进入质谱仪。最典型的就是气相或液相色谱仪通过接口与质谱仪连接。
(3)离子化系统 就是离子化室,通常采用电子轰击电离(EI电离),使用具有一定能量的电子直接作用于样品分子,使其电离。轰击用电子由钨或铼制作的灯丝在高真空中发射,通过灯丝与电离盒之间所加的电压加速。对有机化合物通常选用70 eV的电压。
除了电子轰击的电离方法之外,还有场致电离、化学电离、激光电离等方法。
(4)质量分析系统 质量分析器的作用是将离子源中形成的离子按质荷比的大小不同分开,质量分析器可分为静态分析器和动态分析器两类。
静态分析系统采用稳定不变的电磁场(电磁场随时间改变,只是为了记录质谱而不是分离原理所要求的),并且按照空间位置把不同质荷比(m/Z)的离子分开。属于这一类的仪器有单聚焦磁场分析系统的仪器和双聚焦磁场分析系统的仪器。
动态分析系统采用变化的电磁场,按照时间或空间来区分质量不同的离子。属于这一类的仪器有飞行时间质谱仪、四极质谱仪等。
(5)离子检测系统 通常用电子倍增器检测离子流。电子倍增器中电子通过的时间很短,利用电子倍增器可以实现高灵敏、快速测定。
(6)记录系统 目前质谱仪大多使用计算机作为记录系统,计算机接收离子检测系统提供的信号,进行数据处理,给出质谱图。
2.质谱图
质谱图由横坐标、纵坐标和棒线组成。横坐标标明离子质荷比(m/Z)的数值,纵坐标标明各峰的相对强度(也叫相对丰度),棒线代表质荷比的离子。图谱中最强的一个峰称为基峰,将它的强度定为100。
图1-14为甲基异丁基甲酮的质谱图,从图中可以找到甲基异丁基甲酮裂解所产生的具有不同质荷比(m/Z)的离子碎片的相对强度。
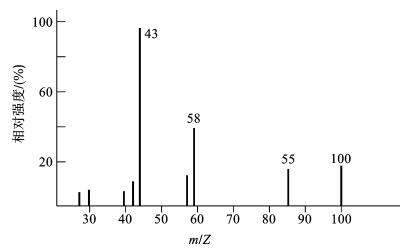
图1-14 甲基异丁基甲酮的质谱图
3.质谱的样品测试
质谱的样品测试比较简单,按照上述的三种进样方法进样即可。
(1)由分子离子峰测定相对分子质量这是质谱的重要用途之一,它比经典的相对分子质量测定方法快而准确,且所需试样量少(一般为0.1mg)。
(2)用质谱图鉴定有机化合物这种情况适用于已知有机化合物。就是将所测得的样品的质谱图与标准图谱库或文献报道的该化合物的图谱进行对比。若两张图谱完全相同,就可以确定该化合物与标准图谱或报道的化合物是同一种物质;若两张谱图不一样,或峰位不一致,则说明两者可能不为同一化合物,或样品有杂质。使用文献上的谱图应当注意测试时裂解条件以及所用仪器类型均应与标准谱图或报道的图谱相同。
目前可检索的标准图谱库有萨德勒(Sadtler)标准MS谱图库、Wiley出版公司的MS数据库、美国国家标准研究所的NIST/EPA/NIH质谱数据库等。
(3)用质谱图推测有机化合物的分子结构这是一项比较复杂的工作,具体步骤可参阅相关教材。