
下载掌阅APP,畅读海量书库
立即打开



锂离子电池自20世纪70年代,第一颗商业化Li/(CF) n 锂一次电池销售以来,已经经过了数十年的发展,对各种正极材料的研究也被广泛展开,Li/(CF) n (1970年)、Li/MnO 2 (1975年)、Li/I 2 (1976年)被相继研发出来。到现在已经商业化的锂一次电池正极材料有:I 2 、MnO 2 、CuO、(CF) n 、SOCl 2 、SO 2 等。
锂离子二次电池正极材料一开始的研究主要集中在可嵌入的化合物中,1972年,EXXO公司设计了以TiS 2 为正极材料的锂离子二次电池,TiS 2 是一种层状半金属,可发生锂的脱嵌反应,具有良好的可逆性,且导电性好,无须添加导电剂,但是材料难以合成且成本较高以及没有解决锂枝晶问题,因此没有投入商业化生产。随后,1980年Goodenough提出了氧化钴锂(LiCoO 2 )可以作为正极材料,但由于传统方法里组装成电池后是满电状态,而氧化钴锂做正极时需要先充电,且主流观点认为高工作电压影响有机电解质的稳定性,因此该正极材料在当时并没有得到足够的重视。1983年,Goodenough又发现了尖晶石型LiMn 2 O 4 可以作为优秀的正极材料,其价格较低,且拥有优良的导电、导锂性能,以及相较于LiCoO 2 更高的安全性。1986年,第一块以MoS 2 为正极材料的锂离子二次电池被加拿大Moli公司推向市场,不过由于锂枝晶并没有被解决导致电池爆炸起火,使该电池设计被终结,Moli公司也因此一蹶不振并最终被收购。在之后1996年Goodenough再次提出橄榄石结构的磷酸铁锂也可以作为锂离子二次电池正极材料,磷酸铁锂具有良好的热稳定性和安全性,且价格低廉、对环境友好等优点。
正极作为锂离子电池四大关键部位之一,正极材料的选择直接决定了锂离子电池的安全性能和电池能否大型化,正极材料的成本也较为高昂,约占锂离子电芯材料成本的1/3左右,同时正极材料也对锂离子电池的电压、容量等多个指标都起到了非常重要的影响。因此寻找一个廉价、高容量、高比能量、安全可靠的正极材料是未来锂离子电池发展的重要挑战。
选择锂离子电池的正极材料时需要考虑几个因素:
1)输出电压:正极材料需要有较高的氧化还原电位,有较高电压的放电平台,以此获得拥有较高电压的电池。
2)高容量:正极材料应该能够允许大量的锂离子进行可逆的嵌入脱出。
3)循环性能好:在锂离子的嵌入脱出过程中,正极材料的结构需要保持稳定,不发生大的变化。
4)大电流放电:正极材料需要具有较高的离子电导率和电子电导率,减少极化现象,满足电池大电流放电的需求。
5)正极材料应该在整个电压范围内具有良好的化学稳定性,不与电解液发生反应。
6)从实用角度来看,材料应该对环境无污染且价格便宜、储量丰富。
锂离子电池的发展已经经历了100多年,现在使用的锂离子电池是在电极材料固态化学方面不断研究和发展的结果。其中,最重要的是开发新型电极材料,并且持续不断地研究其结构-组分-性能-电化学性能之间的关系。这些研究对锂电池的发展起着至关重要的作用。从成本分析来看,正极材料的成本仍然占据了很大的比重。但是,有趣的是目前使用的三种常见的正极材料都是出自美国奥斯汀大学的John B Goodenough课题组,包括层状材料,尖晶石型材料和聚阴离子材料。因此非常有必要来看看这几类材料对锂离子电池的发展演变起着怎样的作用。
1841年,Schauffautl就发现了锂离子可以在石墨材料中脱嵌。随后,在20世纪70年代,脱嵌机理被应用于Li + 与TiS 2 反应中,完美地解释了锂的储存机理。但是由于硫化物的工作电压低,导致了电池的能量密度低。提高材料的能量密度才能进一步提高锂电池的能量密度。Goodenough教授从20世纪80年代开始研究锂离子电池正极材料,根据氧化还原的机理,正极材料的氧化能越低越好,而负极材料的还原能越低越好。研究发现,O 2- :2p 6 的轨道能量比S 2- :3p 6 的能量要低,因此该课题组开始开发氧化物。同时,他们还发现,如果跟过渡金属匹配,过渡金属氧化物的氧化还原电位能够达到较高的水平。基于这个基本的思路,他们分别在1980年发明了层状氧化物LiCoO 2 ,1983年发明了尖晶石型的LiMn 2 O 4 ,1997年发明了聚阴离子材料LiFePO 4 。
LiCoO 2 是一种O3型结构的材料,其中,Li和Co有序地排列在(111)面上。这种有序的结构导致了高的离子迁移率。其中,Co-Co的相互作用导致了较好的电子电导率。基于以上特点,LiCoO 2 克服了硫化物的两个大的弱点:①电极工作电压有很大的提高(>4.0V);②避免了采用金属锂作为电极,使得电池安全性更好。但是它的实际容量大概是140mA·h/g(理论容量为274mA·h/g),伴随着0.5Li + (每化学式)可逆的脱嵌。随后,由于其他的过渡金属,例如Ni、Mn,也具有较好的氧化还原性。因此,这些元素也被单独或者作为掺杂元素被大量的研究,其中最典型的是LiNi 1 -y-z Mn y Co z O 2 (NMC)。近几年由于对于电池能量密度的追求,富锂材料也被发现。
LiMn 2 O 4 是一种磁铁矿石结构的尖晶石的正极材料。跟LiCoO 2 不同,尖晶石型的材料是一种半导体,但是它具有较好的结构稳定性和3D锂迁移通道。由于四面体的Li + 具有较高的位能,所以该材料的氧化还原电压也能达到4.0V。相比于LiCoO 2 ,尖晶石锰酸锂的成本更低。但是它的缺点是Mn的Jahn-Teller效应,影响了该材料的电化学性能。掺杂能够明显改善材料的性能,例如Ni或者F等。
其实,对于聚阴离子化合物的研究从1980年已经开始了,但是它们的工作电压都偏低。有关LiFePO 4 的研究成果是在1997年被发表出来的,它具有两相反应,LiFePO 4 →FePO 4 ,氧化还原电压是3.4V。Li + 在PO 6 六面体和FeO 4 四面体之间,它具有沿着b轴的一维锂离子嵌脱通道扩散,因此,它的离子迁移率要低于金属氧化物。取决于过渡金属元素,聚阴离子材料有可能能将电压提高到5V左右。此外,由于较强的P-O键的存在,材料的结构稳定和安全性相对较高。该材料的缺点是容量低。
作为一个储能元件,电池的最终作用是需要给相关设备提供电能,因此,成本和能量密度是两个最重要的因素。从体积能量密度和质量能量密度上考虑,氧化物具有很大的优势,但是其成本比较高,并且可持续发展性以及安全性也是极其重要的影响因素。这两个方面是氧化物的弱点。因此,从应用领域来看,便携性电子设备使用的小型电池,还是需要进一步提高能量密度和寿命。但是,这并不如电动车领域那么迫切,因为为了使电动车可以跑得更远更安全,需要极大地提高电池的能量密度。高镍材料能够在未来的发展中占据一席之地,因为它具有较高的工作电压,从而提高了材料的能量密度。并且,由于Co的储量分布不均匀,且价格昂贵,因此无钴的电极材料也是极有可能是一种未来的电极材料。如果考虑到安全性能,聚阴离子化合物LiFePO 4 也是热门的正极材料的选择之一,由于其优异的热稳定型,能够保证使用的安全性。开发更高工作电压的聚阴离子正极材料也是未来的发展方向之一,包括是使用钒元素和掺杂氟元素等,都是重要的发展方向。目前,还没有一款电池能够完全满足所有的需求,只能根据各种不同的需求来选择不同的正极材料。从历史发展来看,现在使用的材料被发现和大量研究之前,已经有了很多的不同探索。但是在工业的发展过程中,资源的短缺限制了一些主要材料的发展。因此,科学家和工业界又开始了研究和发展之前被认为不成熟的材料。从最近几年的发展态势来看,结合理论研究、设计和合成具有高能量密度、良好安全性能的正极材料依然被认为是正极材料的开发研究主旋律。
层状氧化物正极材料中,层状钴酸锂(LiCoO
2
)是最早商业化并且也是目前应用最广的材料,这类材料还包括Li
M
O
2
(
M
=Ni,Cr,CoNi,CoNiMn等),它们都是以立方密堆积的氧离子为支撑结构,过渡金属离子和锂离子各自的分层位于氧离子构成的八面体中心位置,具有斜方六面体
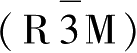 空间群的α-NaFeO
2
型晶体结构,这种结构源于NaCl结构。在这种结构中,锂离子、过渡金属离子和氧离子沿着(111)面做O-Li-O
-M
-O-Li-O
-M
规则排列,层状结构中MnO
2
层间的库仑斥力形成的良好的二维通道可以实现锂离子快速脱嵌,同时
M
与
M
之间以
M
-O
-M
的形式发生相互作用,因此也具有较高的电子电导率。
空间群的α-NaFeO
2
型晶体结构,这种结构源于NaCl结构。在这种结构中,锂离子、过渡金属离子和氧离子沿着(111)面做O-Li-O
-M
-O-Li-O
-M
规则排列,层状结构中MnO
2
层间的库仑斥力形成的良好的二维通道可以实现锂离子快速脱嵌,同时
M
与
M
之间以
M
-O
-M
的形式发生相互作用,因此也具有较高的电子电导率。
目前虽然钴酸锂因为其电位高、适合大规模生产等原因已经被广泛应用,但其价格昂贵,且具有毒性,因此广大科研人员正在不断地寻求其替代物,对镍基正极材料及将过渡金属离子和其他阳离子掺杂的复合材料的研究被广泛地开展。未来的发展方向是使用简单的过渡金属层状氧化物,使每个过渡金属离子至少对应一个可以可逆脱嵌的锂离子,同时可以保持材料的低成本和低毒性,以及足够的安全性。
钴酸锂根据合成方法不同,具有两种不同的结构:①通过800℃以上的固相反应合成的层状结构;②在400℃合成的尖晶石结构 [1] 。但由于尖晶石结构的钴酸锂结构不够稳定,循环性能差,常被人忽略,因此通常意义上所说的钴酸锂均指前者。钴酸锂是最为常用的正极材料,它易于制备且结构稳定,但同时又有资源有限、成本高,具有毒性的缺点。
层状钴酸锂具有斜方六面体
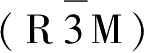 结构,它最早是由Goodenough教授于1980年提出可以用作正极材料。理想的层状LiCoO
2
结构中,锂离子和钴离子各自位于立方紧密堆积氧层中交替的八面体位置,
a
=0.2816nm,
c
=1.4056nm。但实际上由于锂离子、钴离子与氧原子层的作用力不同,因此氧原子的分布并不是理想的密堆积结构,而是有所偏离,呈现出三方对称性。
结构,它最早是由Goodenough教授于1980年提出可以用作正极材料。理想的层状LiCoO
2
结构中,锂离子和钴离子各自位于立方紧密堆积氧层中交替的八面体位置,
a
=0.2816nm,
c
=1.4056nm。但实际上由于锂离子、钴离子与氧原子层的作用力不同,因此氧原子的分布并不是理想的密堆积结构,而是有所偏离,呈现出三方对称性。
充放电过程中,锂离子在键合力强的CoO 2 层间进行二维运动,具有较高的离子电导率,Li + 扩散系数:10 -9 ~10 -7 cm 2 /s。其在3.92V展示出明显的放电平台,工作电压的平均值可以达到3.7V,对于LiCoO 2 来说由于低自旋和八面体位置上稳定的钴离子,使得其在充放电过程不会发生不可逆相变,但是当充电持续进行时,Li 1 -x CoO 2 ( x >0.5)层状O3结构和单斜P3结构相混,此时可能发生不可逆的相变,因此可以看出,钴酸锂充当正极材料时,只有大约50%的锂可以进行可逆的嵌入-脱出,导致钴酸锂的理论比容量虽然达到了274mA·h/g,但实际中的比容量只比理论比的一半(137mA·h/g)稍高,大约为145mA·h/g。当充电进行到 x >0.72时,此时电荷的补充不是通过Co的氧化而是晶格氧的释放,当温度高于50℃,氧的大量释放使得晶格崩塌,同时正极材料和电解液的副反应会释放气体使电池膨胀,使电池安全性变差,完全充电后的LiCoO 2 变为CoO 2 ,为了保证钴酸锂的结构稳定,实际应用中钴酸锂充当正极材料的电池充电电压一般不超过4.4V。
通过在钴酸锂中掺杂若干元素,可以使得材料在 x >0.5时,稳定层状晶格,并且增加LiCo 1 -y M y O 2 的比放电容量 [2,3] ,掺杂元素 M 可以为Al [4,5] 、Mg [6] 、B [7] 、Cr [8] 、Mn等。当 y ≤0.2时,B的掺杂显著改善了电池循环性能,这是因为B的掺杂更有利于锂离子在晶格中的嵌入脱出,并且抑制了Li 0.5 CoO 2 的Verwey相变相关的一阶结构相变,其作用还包括降低极化作用以及减少电解液的分解。层状LiCo 0.7 Ni 0.3 O 2 、LiCo 0.7 Al 0.3 O 2 的电化学特征与LiCoO 2 类似,Al基和Ni基的掺杂使得材料电化学性能有所提高,显示了良好的循环性能,而且长循环下的容量保持效率并不是以牺牲初始的可逆容量为代价的,采用Al掺杂的因素有几个,包括其价格便宜、毒性低、密度小,同时α-LiAlO 2 的结构与LiCoO 2 结构类似且铝离子与钴离子的半径基本相同,Al的掺杂也可以提高电压稳定结构,Ni和Mg的掺杂也可以提高结构的稳定性和循环寿命,Mn的掺杂不仅可以使电池可逆容量增加,也可以提高电池的工作电压。但是LiCo 0.7 Cr 0.3 O 2 电池却显示出严重的容量衰减 [9] ,这主要由于取代造成了结构扭曲。
为了改善钴酸锂的电化学性能,也常常采用对钴酸锂表面包覆其他材料的方法,包覆材料主要为无机氧化物,例如MgO,Al 2 O 3 ,LiMn 2 O 4 ,AlPO 4 ,SnO 2 等,不同材料的包覆具有不同的作用,MgO的包覆可以提高结构的稳定性 [9] ,无定形Al 2 O 3 的包覆可以防止Co的溶解,稳定钴酸锂层状结构,提高循环性能。表面的LiMn 2 O 4 或AlPO 4 包覆可以使钴酸锂的热分解温度从185℃提高到225℃,而且循环性能也有显著的提高。同时也可以对钴酸锂进行碳包覆,在碳包覆之后,钴酸锂材料在大电流下的充放电性能得到了显著的提高,这主要是因为碳的包覆提高了电子的导电性。
镍酸锂与钴酸锂同构,都是 α-NaFeO
2
型层状结构,属于
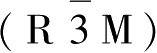 空间群,
a
=0.2886nm,
c
=1.4214nm,
c/a
=4.928。与钴酸锂一样,由于Ni
3+
/Ni
4+
的耦合使得LiNiO
2
具有较高的对锂电位,约为4V,Li
+
扩散系数为2×10
-7
cm
2
/s。相较于钴酸锂,镍酸锂的实际比容量更高,且对于钴,镍在地壳中的含量更高,成本低且毒性低,因此被广泛研究。希望镍酸锂可以替代钴酸锂,但是镍酸锂存在的一些问题阻碍了其实际应用。首先是由于Ni
2+
较难氧化为Ni
3+
,因此通常所合成的LiNiO
2
中部分Ni
3+
被Ni
2+
取代,为了保持电荷平衡,一部分Ni
2+
会占据Li
+
所在的位置,因此NiO
2
层会形成一个局部的三维结构,这阻碍了锂离子的扩散,降低了充放电效率;同时Ni
2+
半径略小于Li
+
,被氧化成Ni
3+
半径更小,在脱锂过程中会使得层间局部结构塌陷,使得该镍离子周围的6个锂位难以再次嵌入,造成材料的容量损失,使电池的循环性能下降。此外,低自旋的Ni
3+
(d
7
)电子构型存在Jahn-Teller效应
[10]
,使得
z
轴方向的键长增加,LiNiO
2
在充放电过程中
z
轴方向上反复膨胀收缩,使得电导率降低,电极性能恶化。LiNiO
2
的热稳定性在同条件下相比于LiCoO
2
也更差,其热分解温度在200℃附近,且放热量多,这是因为充电后期的四价镍处于高氧化态,其强氧化性不仅会氧化分解电解质还会腐蚀集流体,放出热量以及氧气
[11]
,而且自身在一定温度下容易放热分解,当聚集到一定程度时,就可能会发生爆炸。
空间群,
a
=0.2886nm,
c
=1.4214nm,
c/a
=4.928。与钴酸锂一样,由于Ni
3+
/Ni
4+
的耦合使得LiNiO
2
具有较高的对锂电位,约为4V,Li
+
扩散系数为2×10
-7
cm
2
/s。相较于钴酸锂,镍酸锂的实际比容量更高,且对于钴,镍在地壳中的含量更高,成本低且毒性低,因此被广泛研究。希望镍酸锂可以替代钴酸锂,但是镍酸锂存在的一些问题阻碍了其实际应用。首先是由于Ni
2+
较难氧化为Ni
3+
,因此通常所合成的LiNiO
2
中部分Ni
3+
被Ni
2+
取代,为了保持电荷平衡,一部分Ni
2+
会占据Li
+
所在的位置,因此NiO
2
层会形成一个局部的三维结构,这阻碍了锂离子的扩散,降低了充放电效率;同时Ni
2+
半径略小于Li
+
,被氧化成Ni
3+
半径更小,在脱锂过程中会使得层间局部结构塌陷,使得该镍离子周围的6个锂位难以再次嵌入,造成材料的容量损失,使电池的循环性能下降。此外,低自旋的Ni
3+
(d
7
)电子构型存在Jahn-Teller效应
[10]
,使得
z
轴方向的键长增加,LiNiO
2
在充放电过程中
z
轴方向上反复膨胀收缩,使得电导率降低,电极性能恶化。LiNiO
2
的热稳定性在同条件下相比于LiCoO
2
也更差,其热分解温度在200℃附近,且放热量多,这是因为充电后期的四价镍处于高氧化态,其强氧化性不仅会氧化分解电解质还会腐蚀集流体,放出热量以及氧气
[11]
,而且自身在一定温度下容易放热分解,当聚集到一定程度时,就可能会发生爆炸。
为了解决理想LiNiO 2 难以批量制备,稳定性差,充放电过程存在相变的问题,人们采取了许多办法想要将其改性,改性的方向主要有:
1)防止LiNiO 2 与电解液直接接触,减少副反应。
2)降低循环过程中的产热量。
3)抑制相变,提高结构稳定性。
4)减少界面阻抗的增加。
5)提高可逆容量,降低不可逆容量比例。
为此采取的方法主要集中在采用溶胶-凝胶法,加入掺杂元素和进行包覆。
溶胶-凝胶法主要通过将金属阳离子和锂离子构成混合盐溶解于含络合剂的水溶液中,通过溶胶-凝胶化反应使金属阳离子和锂离子均匀分布在凝胶化树脂网络中,充分干燥后经高温烧结过程可以制备出组分均匀的材料,通过溶胶凝胶法制备出的镍酸锂热稳定性可以提高到400℃以上,初始容量在150mA·h/g以上。
掺杂元素的目的是为了提高镍酸锂晶体结构在循环中的稳定性,可掺杂的元素有单一元素掺杂,如Li、F、Na、Mg、Al、Ca、Ti、Mn、Co等;多种元素掺杂,如Li和Co、Co和F、Co和Mg、Co和Al、Co和Mn等。Li的掺杂一般而言称之为过量锂的加入,但是一般来说不利于电化学性能的提高。F的单独掺杂主要是取代部分氧原子,使部分二价镍占据锂离子的位置,增加氧离子的无序性,由于内部阻抗的减少,电化学的性能反而得到了提高。Na的掺杂是为了取代锂,生成Li
x
Na
1
-x
NiO
2
,且随着
x
的不同,相的状态也会发生变化,
x
=0.0时为单斜相(C2/m);当0.13<
x
<0.15时,为第一种菱形相
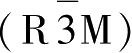 ,在这种相中,锂离子和钠离子的位置没有无序的二价镍离子,并且不会出现因相邻镍层位置变化时而产生的杨-泰勒效应;当
x
=1时,为第二种菱形相
,在这种相中,锂离子和钠离子的位置没有无序的二价镍离子,并且不会出现因相邻镍层位置变化时而产生的杨-泰勒效应;当
x
=1时,为第二种菱形相
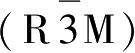 。第一种菱形相在作为正极材料时具有良好的前景。镁离子掺杂时主要跟量有关,量较少时,主要是二价镍被取代,因此循环性能较好;但是过量时,镁离子开始取代四价镍,将会得到明显不同的电化学性能。
。第一种菱形相在作为正极材料时具有良好的前景。镁离子掺杂时主要跟量有关,量较少时,主要是二价镍被取代,因此循环性能较好;但是过量时,镁离子开始取代四价镍,将会得到明显不同的电化学性能。
包覆同钴酸锂一样,对镍酸锂进行表面包覆也可以改善其性能,可以对其进行包覆的材料种类非常多,如ZrO 2 ,Li 2 O·2B 2 O 3 玻璃体,MgO等,主要作用有以下四点:
1)防止镍酸锂与电解液之间的直接接触,减少副反应的发生。
2)提高了表面性能,可以减少循环过程中的产热量。
3)抑制相变,提高结构稳定性。
4)减少了界面阻抗的增加。
层状结构的LiMnO 2 由于热力学上不稳定,因此合成难度比较高,层状的正交LiMnO 2 的结构与层状钴酸锂有点不同,属于岩盐结构,其氧原子堆积方式为扭变四方密堆结构,交替的锂离子层和锰离子层发生褶皱。因此尽管同为层状结构,但是LiMnO 2 的氧离子层与密堆氧平面并不平行,其对称性要差于钴酸锂,其空间点群为Pmnm。LiMnO 2 的理论容量为285mA·h/g,其在2.0~4.5V充电时,初始容量可以达到200mA·h/g,但是由于脱出锂后的Li 1 -x MnO 2 结构很不稳定,极容易转变成较稳定的尖晶石结构Li 1 -x Mn 2 O 4 。
三元材料Li[Ni x Co y Mn z ]O 2 融合了高容量LiNiO 2 、热稳定性好和价格便宜的LiMnO 2 、电化学性能稳定LiCoO 2 的优点,展现了优秀的电化学性能。其从组成来看应该归于镍酸锂,但是它是从层状氧化锰锂发展而来的,主要有Li[Ni 1/3 Co 1/3 Mn 1/3 ]O 2 ,Li[Ni 0.4 Co 0.4 Mn 0.2 ]O 2 ,Li[Ni 0.5 Co 0.2 Mn 0.3 ]O 2 ,Li[Ni 0.8 Co 0.1 Mn 0.1 ]O 2 ,为了避免Mn 3+ 的Jahn-Teller效应,Ni离子和Mn离子的量最好保持相等,其都是从Li[Ni 1/3 Co 1/3 Mn 1/3 ]O 2 发展而来,这里重点描述Li[Ni 1/3 Co 1/3 Mn 1/3 ]O 2 。
Li[Ni
1/3
Co
1/3
Mn
1/3
]O
2
具有单一的α-NaFeO
2
型层状岩盐结构,空间点群
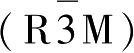 ,
a
=0.4904nm,
c
=1.3884nm。在Li[Ni
1/3
Co
1/3
Mn
1/3
]O
2
中过渡金属元素Co、Ni、Mn分别以+3、+2、+4价态存在,锂离子占据岩盐结构的3a位,过渡金属离子占据3b位,氧离子占据6c位,其中的Co的电子结构与钴酸锂(LiCoO
2
)中的Co相同,但是Ni和Mn的电子结构却与LiNiO
2
和LiMnO
2
不同。Li
1
-x
[Ni
1/3
Co
1/3
Mn
1/3
]O
2
中,通常认为镍离子参与充放电的全程,其在0≤
x
≤1/3范围内发生Ni
2+
/Ni
3+
的氧化还原反应,对应3.9V左右的放电平台;在1/3≤
x
≤2/3范围内发生Ni
3+
/Ni
4+
的氧化还原反应,对应3.9~4.1V的放电平台,Co
3+
在充电末期被激活;即在2/3≤
x
≤1时发生Co
3+
/Co
4+
的氧化还原反应,与上述的两个放电平台都有关系。Mn在整个充放电过程中不发生氧化还原反应,只是作为一种结构物质,其通过八面体位置的晶体场稳定化能来提供整个晶体结构的稳定性,不会出现层状结构向尖晶石结构的转变。
,
a
=0.4904nm,
c
=1.3884nm。在Li[Ni
1/3
Co
1/3
Mn
1/3
]O
2
中过渡金属元素Co、Ni、Mn分别以+3、+2、+4价态存在,锂离子占据岩盐结构的3a位,过渡金属离子占据3b位,氧离子占据6c位,其中的Co的电子结构与钴酸锂(LiCoO
2
)中的Co相同,但是Ni和Mn的电子结构却与LiNiO
2
和LiMnO
2
不同。Li
1
-x
[Ni
1/3
Co
1/3
Mn
1/3
]O
2
中,通常认为镍离子参与充放电的全程,其在0≤
x
≤1/3范围内发生Ni
2+
/Ni
3+
的氧化还原反应,对应3.9V左右的放电平台;在1/3≤
x
≤2/3范围内发生Ni
3+
/Ni
4+
的氧化还原反应,对应3.9~4.1V的放电平台,Co
3+
在充电末期被激活;即在2/3≤
x
≤1时发生Co
3+
/Co
4+
的氧化还原反应,与上述的两个放电平台都有关系。Mn在整个充放电过程中不发生氧化还原反应,只是作为一种结构物质,其通过八面体位置的晶体场稳定化能来提供整个晶体结构的稳定性,不会出现层状结构向尖晶石结构的转变。
LiNiO 2 在3.0~4.3V范围内有三对可逆的氧化还原峰,而Li[Ni 1/3 Co 1/3 Mn 1/3 ]O 2 只有一对,可以认为其中的LiNiO 2 组分在充放电过程中的多次相变得到了很好的抑制。Li 1 -x [Ni 1/3 Co 1/3 Mn 1/3 ]O 2 在充放电过程中还是会发生结构的变化,其主要表现在晶格参数 c 随着锂含量的减少而增大。但当(1 -x )<0.35时,由于氧的产生而减小,晶格参数 a 的变化正好相反 [12] ,但总的来说,这种材料的体积变化很小。Li[Ni 1/3 Co 1/3 Mn 1/3 ]O 2 稳定的结构使得相应的电池表现出长寿命和高安全性。
由于与钴酸锂相近的容量,但是拥有更高的安全性和更低的成本,因此Li 1 -x [Ni 1/3 Co 1/3 Mn 1/3 ]O 2 是钴酸锂一种很好的替代品。实际上到目前为止,Li 1 -x [Ni 1/3 Co 1/3 Mn 1/3 ]O 2 材料内部的精细结构以及层间过渡金属原子的排布和作用机理还未形成统一完整的理论,限制了其结构理论在新型锂离子电池材料设计和制备中的指导作用,这是研究者共同探索的方向之一。与此同时对各种不同材料的复合的研究也在不断进行,其中一个想法是增加Ni含量以提高其容量的同时又不会使其丧失原有的优势。
三元层状正极材料的合成方法主要有4种,分别是固相烧结法、先驱体共沉淀法、喷雾热解法、溶胶-凝胶法。
1.固相烧结法
高温固相烧结法是制备粉体材料常用的方法,其工艺简单,设备易于构建。但是这种方法的原料混合不均匀以及材料的粒径分布不均匀。
将金属阳离子盐与锂盐采用研磨或者球磨的方法均匀混合,在特定氛围下经高温烧结得到正极材料。常用的金属阳离子锂盐有金属氧化物、乙酸盐、碳酸盐、氢氧化物等。
2.先驱体共沉淀法
先驱体共沉淀是将可溶性金属阳离子盐溶于去离子水中,通过氢氧根或碳酸根等沉淀剂形成两种或两种以上的阳离子的沉淀,再将共沉淀产物与化学计量比锂盐共混后经高温烧结得到产物,这种方法有利于提高金属阳离子混合程度。
在先驱体共沉淀法中,反应体系的pH值和反应温度对于产物的粒径、形貌和元素比分布等有着很大的影响,烧结温度和时间对材料的结晶性能和充放电特性有很大的影响。
该方法制备正极材料的前驱体主要包括两类:一类是碳酸盐共沉淀,一类是氢氧化物共沉淀。前者制备的前驱体在后续热处理过程中存在很大的失重,材料易形成疏散结构,振实密度较低。后者通过调节体系的pH值、搅拌速度、络合剂的用量,可制备粒径较小、分布均一的类球形前驱体,与LiOH烧结,得到振实密度高的正极材料。
3.喷雾热解法
喷雾热解法是能够连续制备超细粉体材料的重要方法,该方法能够得到高振实密度的粉体,具有制备时间短、形貌易于控制、中间环节少等优点。
具体过程如下:将金属阳离子溶解于溶剂中,利用超声波喷雾器将混合溶液离子化,然后喷入反应装置中反应制备得到的先驱体,经与锂盐混合后在高温状态下经一定时间烧结可制备正极材料。
实验表明,喷雾降解法有利于得到尺寸均匀的球形产物,提高材料倍率性能和循环稳定性。
4.溶胶-凝胶法
溶胶-凝胶法能通过螯合剂充分分散多种金属阳离子,并通过有机物在烧结过程中辅助得到结晶均匀的正极材料,材料混合均匀,晶粒尺寸均匀。
溶胶-凝胶法主要通过将金属阳离子和锂离子均匀分散在凝胶化树脂网络中,充分干燥后经过高温烧结过程制备得到组分均匀的正极材料。
尖晶石型锰酸锂(LiMn 2 O 4 )充当正极材料最早是由Goodenough教授等在1983年提出的,其具有价格低、稳定、导电性好、导锂性好等优点,而且其分解温度高,且氧化性远低于钴酸锂,因此在出现短路和过充时可以避免燃烧爆炸,具有很好的安全性。不过随着研究的开展人们发现其在循环过程中由于Jahn-Teller畸变、锰在电解液中的溶解等问题会造成容量的衰减,因此采取诸如掺杂改性等多种方法来改善其性能。
尖晶石结构LiMn
2
O
4
中氧按照ABCABC立方密堆积。LiMn
2
O
4
具有四方对称性,属于
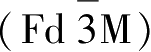 空间群,晶格常数
a
=0.8245nm。一个晶胞中含有8个锂原子,16个锰原子,32个氧原子,锰原子中Mn
3+
/Mn
4+
各占50%。在该结构中,通过三维连接的共面八面体为充放电过程中锂离子的迁移提供途径,由于大量锰的存在即使在脱锂状态下,氧原子仍然能保持理想的立方密堆积状态。
空间群,晶格常数
a
=0.8245nm。一个晶胞中含有8个锂原子,16个锰原子,32个氧原子,锰原子中Mn
3+
/Mn
4+
各占50%。在该结构中,通过三维连接的共面八面体为充放电过程中锂离子的迁移提供途径,由于大量锰的存在即使在脱锂状态下,氧原子仍然能保持理想的立方密堆积状态。
LiMn 2 O 4 在充放电过程中表现出4V和3V两个电压平台 [13] ,当Li 1 -x Mn 2 O 4 在0≤1 -x ≤1时,在四面体8a位置的锂离子在4V左右发生脱嵌;当Li 1+ x Mn 2 O 4 在1≤1+ x ≤2时,在八面体16c位置的锂离子在3V左右发生脱嵌,同时还伴随着立方相LiMn 2 O 4 和四方相Li 2 Mn 2 O 4 之间的相变,其同样的Mn 3+ /Mn 4+ 的氧化还原反应却有1V的电势差的主要原因是立方相8a位置的锂离子和四方相16c位置的锂离子的能隙引起的 [14] 。在放电初始阶段,Mn在LiMn 2 O 4 中的平均化合价为3.5,因为8a四面体位置接近占50%八面体的16c位置,因此锂离子可沿8a→16c→8a→16c→8a的路径迁移。同时Mn 2 O 4 尖晶石结构保持不变,为锂离子提供了一条短的扩散路径,因此拥有高的离子电导率。但随着放电进行,过量的锂离子嵌入,Mn从3.5价还原到3.0价,由此引起的Jahn-Teller形变,使得沿 c 轴的Mn—O键变长, a 轴和 b 轴的Mn—O键变短,并导致Li 2 Mn 2 O 4 表现出宏观四方对称性,从立方相到四方相的转变中,c/a比例变化高达16%,单元晶胞体积增加了6.5%。这足以导致表面的尖晶石粒子发生破裂,因此在这种情况下,即使两个锂离子可以进行可逆的嵌入脱出,但大的体积变化会对材料的结构完整性造成破坏,从而引起电池容量的快速衰减。综上所述,LiMn 2 O 4 只能用于4V区域,其实际容量仅为120mA·h/g。
然而即使在4V区域内,LiMn 2 O 4 仍然会表现出容量的衰减,造成容量衰减的原因包括Mn在电解液中的溶解,其可以归结于粒子表面的Mn 3+ 发生如下歧化反应 [15] :2Mn 3+ (固)→Mn 4+ (固)+Mn 2+ (溶液)。同时溶解的锰离子还会破坏锂离子在负极的电沉积或者成为电解液分解的催化剂,在高温下,这种催化反应加强,使得容量下降更加明显。此外还包括充电尽头,Mn 4+ 的高氧化性在有机溶剂中的不稳定性等原因。
可以总结出尖晶石型锰酸锂相较于钴酸锂有3个优点,①Mn元素在自然界中的存量比钴和镍更加丰富;②Mn毒性较低;③可以形成较高的电池电压。但是LiMn 2 O 4 也存在3个缺点:①难以制备高质量的样品;②尽管尖晶石锰酸锂在4V域内不会产生Jahn-Teller形变,但由于局部放电,锰酸锂颗粒的表面可以观察到四方相Li 2 Mn 2 O 4 的形成;③LiMn 2 O 4 在每一个锰对应的锂插入量多于1时转变为四方相。
从上面可以看到,尖晶石结构的LiMn 2 O 4 存在着容量容易衰减的问题,同时其电导率也相对较低,因此人们采取了一些方法对其进行改性:
1)对锰酸锂LiMn 2 O 4 进行表面修饰。对于LiMn 2 O 4 来说,锰在电解液中的溶解是个主要问题,以及LiMn 2 O 4 与电解质之间的反应随着温度升高而加剧。在这种情况下,可以在LiMn 2 O 4 表面包覆Al 2 O 3 、ZrO 2 、ZnO、SiO 2 、Bi 2 O 3 等金属氧化物,在减少Mn 2+ 溶解的同时可以避免电极遭受高温的侵害。
2)通过增大尖晶石粒径的减小其比表面积,以此来减少Mn 2+ 溶解的方法也被采用,但是这种方法是有限度的,因为其在减少Mn 2+ 溶解的同时也使得锂离子的扩散变得困难,限制了尖晶石的电化学性能。
3)除此之外,对LiMn 2 O 4 进行掺杂的方法也被广泛应用,可以掺杂的阳离子种类很多,如Li、B、Mg、Al、Ti、Gr、Fe等。但是对其掺杂阳离子会对LiMn 2 O 4 的电化学性能产生有利和不利两种结果:
有利的掺杂主要起到如下的作用:①提高锰的价态,从而抑制Jahn-Teller效应,例如Li、Mg、Zn;②稳定尖晶石LiMn 2 O 4 框架结构,减少充放电过程中结构的变化,如Gr、Co、Ni;③提高导电性,有利于锂的可逆嵌入脱嵌;④减少比表面积,相应地减少活性物质与电解液之间的接触,降低电解质与电极的分解反应速率和自放电速率,如Co;⑤提高尖晶石结构的晶格参数,促进锂离子扩散系数的提高,如Cr。
不利的掺杂在于如下的3个原因:①降低锰的价态,从而加剧Jahn-Teller效应,如Ti;②生成杂相,破坏尖晶石框架结构的稳定性,不利于锂的嵌入脱嵌,如B、Al;③导致尖晶石LiMn 2 O 4 的晶胞单元体积减小,抑制锂的迁移,如Al。
除了掺杂阳离子外,还可以掺杂阴离子,如O、F、I、S、Se等,以及两种以上离子共同掺杂。其中,F的掺杂可以降低锰在有机溶剂中的溶解度,提高较高温度下的储存稳定性。I、S由于其半径比O大,在Li嵌入时形变小,在循环过程中可以保持结构的稳定性,克服材料在3V时发生的Jahn-Teller效应。Al和F的共同掺杂不仅可以提高尖晶石结构在室温下的稳定性,而且在较高温度下的稳定性也有所提高。Ni和Li的共同掺杂不仅在室温和高温(60℃)下具有良好的循环性能,在4C倍率下也具有良好的循环性能。
通过采用过渡金属取代部分锰的方法得到的尖晶石材料Li M y Mn 2 -y O 4 ( M =Ni,Co,Fe,Cr等) [16] ,在5V左右对金属锂呈现出了高电压平台 [17,18] ,其中LiNi 0.5 Mn 1.5 O 4 具有较高的放电比容量和良好的循环性能,且在4.7V左右存在稳定的充放电平台,具有良好的应用前景。其属于立方晶系,与尖晶石LiMn 2 O 4 具有相同的晶体结构,其4.7V的充放电平台来自于二价镍和四价镍之间的氧化还原变化,而4.1V左右的充放电平台归因于三价锰与四价锰之间的氧化还原。
尖晶石锰酸锂(LiMn 2 O 4 )的常规制备方法通常采用固体反应法,将锂的氢氧化物(或碳酸盐,硝酸盐)和锰的氧化物(或氢氧化物,碳酸盐)混合,在高温如700~900℃下煅烧数小时,即可得到尖晶石LiMn 2 O 4 。
该法制备的材料通常有以下的缺点:物相不均匀,晶粒无规则,晶界尺寸大,且粒度分布范围宽,煅烧时间长。
除此之外,还有的改性制备方法,如改性的共沉淀方法、配体交换法、微波加热法、燃烧法以及一些非经典方法,如脉冲激光沉淀法、等离子提升化学气相沉积法、射频磁旋喷射法等。改性的共沉淀法,比如将硬脂酸加入到羟氧化四甲基铵溶液中,可明显改进尖晶石锰酸锂的大电流性能。微波法是将原料如电解MnO 2 和Li 2 CO 3 混合,在微波合成反应腔中空气气氛下700~800℃进行反应,合成的尖晶石锰酸锂初始容量为140mA·h/g。
磷酸盐Li M PO 4 ( M =Mn、Fe、Co、Ni)为有序的橄榄石结构,其属于正交晶系,空间群为Pmnb [19] ,O以微变形的六方密堆积,P占据四面体空隙,Li和 M 交替占据a-c面上的八面体空隙,形成一个具有一维锂离子通道的三维框架结构。
在上述的磷酸盐中,因为Fe的储量丰富、价格便宜且对环境无毒、可逆性好,因此被广泛开展研究。LiFePO 4 最早由Goodenough教授于1997年提出可应用于正极材料的特性。其具有以下的九个优点:
1)LiFePO 4 电池的标称电压是3.2V(稳定的放电平台)、终止充电电压是3.6V、终止放电电压是2.0V。
2)比容量大,高效率输出:标准放电为2~5V、连续高电流放电可达10C,瞬间脉冲放电(10s)可达20C。
3)工作温度范围较广(-20~+75℃),高温时性能良好:外部温度65℃时内部温度则高达95℃,电池放电结束时温度可达160℃,电池内部结构安全、完好。
4)即使电池内部或外部受到损坏,电池也不燃烧、不爆炸、安全性能最好。
5)极好的循环寿命,经500次循环,其放电容量仍大于95%;实验室制备的磷酸铁锂单体电池在进行1C的循环测试时,循环寿命高达2000次。
6)过放电到0V也无损坏,0电压存放7天后电池无泄漏,性能良好,容量为100%;存放30天后,无泄漏、性能良好,容量为98%;存放30天后的电池再做3次充放电循环,容量又恢复到100%。
7)可快速充电,自放电少,无记忆效应;可大电流2C快速充放电,在专用充电器下,1.5C充电40min即可使电池充满,启动电流可达2C。
8)低成本。
9)对环境无污染。
同时也存在了一些缺点:
1)导电性差。目前在实际生产过程中通过在前驱体添加有机碳源和高价金属离子联合掺杂的办法来改善材料的导电性,研究表明,磷酸铁锂的电导率提高了7个数量级,使磷酸铁锂具备了和钴酸锂相近的导电特性。
2)锂离子扩散速率慢。目前采取的解决方案主要有纳米化LiFePO 4 晶粒,从而减少锂离子在晶粒中的扩散距离;再者就是改善掺杂改善离子的扩散通道。后一种方法看起来效果并不明显;纳米化已经有较多的研究,但是难以应用到实际工业生产中。
3)振实密度较低。一般只能到达0.8~1.3g/cm 3 ,低的振实密度可以说是磷酸铁锂的最大缺点。但这一缺点在动力电池方面不会突出,因此,磷酸铁锂主要用来制作动力电池。
4)磷酸铁锂电池低温性能差。在0℃时的容量保持率约60%~70%,-10℃时为40%~55%,-20℃时为20%~40%,这样的低温性能显然不能满足动力电源的使用要求。当前一些工厂通过改进电解液体系、改进正极配方、改进材料性能和改善电芯结构设计等使磷酸铁锂的低温性能有所提高。
所以下面主要介绍磷酸铁锂。
磷酸铁锂(LiFePO 4 )的结构为橄榄石结构的空间点群为Pmnb,晶格参数 a =0.6008nm, b =1.0334nm, c =0.4693nm,晶胞体积为0.2914nm 3 。它包含一个扭曲的六方密堆积氧框架,Li和Fe各占一半八面体位置,P占据1/8的四面体位置 [20] ,其完全脱锂后生成的FePO 4 具有相似的晶体结构,同属Pmnb空间点群, a =0.5792nm, b =0.9821nm, c =0.4788nm,晶胞体积为0.2724nm 3 。由于在LiFePO 4 晶格中,处于两个Fe-O层之间的P-O四面体链起到了支撑结构的作用,因此在脱锂过程中晶胞体积变化很小;又因为脱锂后产物结构与LiFePO 4 类似,因此LiFePO 4 具有非常好的循环性能 [21] 。除此之外,O与P之间很强的共价键形成四面体聚阴离子,即使在全充状态下,O也很难脱出。因此LiFePO 4 在充电过程中不会有过氧离子的出现,避免了其与有机溶剂发生反应,使LiFePO 4 材料的安全性很高。
此外,相较于LiCoO 2 的充电状态,CoO 2 开始分解产生氧气的温度为240℃,放出热量约为1000J/g,LiFePO 4 的充电状态FePO 4 在210~410℃范围内产生的热量仅为210J/g,拥有良好的热稳定性。
不过由于相邻的FeO 6 共顶点链接,没有共边的FeO 6 八面体网络,造成其电子导电率较低。而且PO 4 四面体位于FeO 6 层之间,一定程度上阻碍了Li + 的扩散运动,使Li + 脱嵌困难。这使得磷酸铁锂的电子电导率和锂离子电导率都很低。
在室温下充放电,其容量会随着电流密度变大而减小,当减少电流密度时,容量又恢复到以前的水平,这是因为随着表面锂的脱嵌,会形成FePO 4 与LiFePO 4 两相界面。随着锂的不断脱嵌,界面逐渐变小,当小到一定程度后,锂通过该界面的迁移速率无法满足该电流强度的需求,电化学行为受到了锂离子扩散的影响;且由于生成的FePO 4 电子电导率和离子电导率很差,在形成的两相结构中,中心的FePO 4 不能够被充分利用,在大电流下FePO 4 的实际利用率明显降低,因此其只适用于小电流放电。
同时因为锂的扩散是通过一维通道进行的,因此会受到材料缺陷的影响。当锂离子通道被如铁离子所占据,就会堵塞锂离子的迁移通道,限制锂离子迁移,且由于铁离子的迁移能力差,这一阻塞的锂离子通道会一直保持非活性状态。
为了解决LiFePO 4 的材料振实密度低,锂离子扩散系数小,电子电导率低等问题,一般对其采用碳包覆、掺杂、纳米化等方法对其改性。
在LiFePO 4 表面进行碳包覆可以有效提高其导电性 [22] ,改善电化学性能。碳的主要作用包括有机物在高温惰性气体的条件下分解成碳,可以从表面增加它的导电性,产生的碳微粒达到纳米级别可以细化产物晶粒,扩大导电面积,有利于锂离子的扩散,还起到了还原剂的作用,避免生成三价铁离子,同时包覆碳增强了粒子之间的导电性,减少了电极充放电过程中的极化等。
对LiFePO 4 的掺杂包括掺杂金属粒子和金属离子,掺杂金属粒子主要是为了增强LiFePO 4 的导电性,如在其中加入少量导电金属颗粒是提高LiFePO 4 电子导电率和容量的另一途径,而金属离子的掺杂是通过制作材料晶格缺陷从而从材料内部出发提高电子导电性。
对LiFePO 4 纳米化是由于其锂离子扩散系数小,当粒子颗粒较大时,限制了大电流性能。
制备LiFePO 4 的方法比较多,主要有固相合成法、碳热还原法、溶胶-凝胶法、模板法、共沉淀法、水热法、溶剂热法、脉冲激光沉积法、微波加热法等,这里简单介绍一下三种常用方法等。
1)固相合成法是最早用于LiFePO 4 合成的方法,也是目前制备LiFePO 4 最常用、最成熟的方法。固相合成用的铁源一般为Fe(C 2 O 4 )·2H 2 O或Fe(OOCCH 3 ) 2 ,锂源一般为Li 2 CO 3 、LiOH·H 2 O或CH 3 COOLi·2H 2 O,而磷源为(NH 4 ) 2 HPO 4 或NH 4 H 2 PO 4 ,将原料按一定比例均匀混合,在惰性气体保护下于300~350℃预烧5~12h以分解磷酸盐、草酸盐或乙酸盐,然后在550~700℃煅烧10~20h。该方法的关键之一是将原料混合均匀,因此必须在热处理之前对原料进行机械研磨。该方法设备和工艺简单,制备条件容易控制,适合工业化生产,但是存在物相不均匀,产物颗粒大、粒度分布范围宽等缺点。
2)碳热还原法以廉价的三价铁作为铁源,在其中混合过量的碳,利用碳在高温下将三价铁还原到二价铁,合理解决了原料混合加工过程中可能引发的氧化还原反应,使得制备过程更合理,同时也改善了材料的导电性,但是该方法的缺点是合成条件苛刻,合成时间长。
3)溶胶-凝胶法制备LiFePO 4 的流程为先在LiOH和Fe(NO 3 ) 3 中加入还原剂,然后加入磷酸,通过氨水调节pH值,将60℃下获得的凝胶进行热处理,便得到了LiFePO 4 。这个方法具有前驱体溶液化学均匀性好、凝胶热处理温度低、粉体颗粒粒径小且分布均匀、粉体焙烧性能好、反应过程易于控制、设备简单等优点,但是干燥收缩大,工业化生产难度较大,合成周期长,同时用到大量有机溶剂,造成了成本的提高和原料的浪费。
过渡族金属元素中,钒的价格低于钴、锰等,其拥有从V 2+ 、V 3+ 、V 4+ 、V 5+ 等多种价态,可以形成多种氧化物,如VO 2 、V 2 O 5 、V 6 O 13 、V 4 O 9 、V 3 O 7 等,其有三种稳定的氧化态V 3+ 、V 4+ 、V 5+ ,可以形成氧密堆分布,因此钒的氧化物为锂二次电池有潜力的候选者。其中V 2 O 5 、VO 2 、V 2 O 5 、LiV 3 O 8 已经被证明可以用于插锂反应,它还能与锂形成多种复合氧化物Li—V—O,与Li—Co—O化合物一样,存在着两种结构:层状结构和尖晶石结构。层状化合物有LiVO 2 、Li x V 2 O 4 和Li 1+ x V 3 O 8 ,尖晶石有正常尖晶石Li[V 2 ]O 4 和反尖晶石V[Li M ]O 4 ( M =Ni、Co)两种。
α-V
2
O
5
为层状结构,属于正交晶胞结构和Pmnm空间群,晶格参数为
a
=1.1510nm,
b
=0.3563nm,
c
=4.369nm
[23,24]
。在钒的氧化物体系中,拥有最高的理论容量442mA·h/g,1mol该物质可以嵌入3mol Li形成Li
3
V
2
O
5
的计量化合物。α-V
2
O
5
层状结构中,氧为扭变密堆分布,钒离子与五个氧原子键合较强,形成四方棱锥络合,随着Li在V
2
O
5
嵌入的
x
值的增加会形成几种Li
x
V
2
O
5
的相(α、β、δ、γ、ω),其相与嵌入Li的
x
值紧密相关,
x
=1时得到δ-Li
x
V
2
O
5
。在0≤
x
≤1时,锂的嵌入脱出时可逆的。
x
=2时,得到γ-Li
x
V
2
O
5
,尽管γ-Li
x
V
2
O
5
不能再回到初始相,但是当充到高电压时,所有的锂均能发生脱嵌。当
x
>2时,结构发生明显变化,钒原子从原来的位置迁移到邻近的空八面体位置,得到岩盐结构的
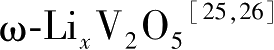 ,钒离子在八面体位置发生无规分布,并发现有氧化锂生成。V
2
O
5
对过充很敏感,随着嵌入的锂量增加,电荷转移变得不可逆。
,钒离子在八面体位置发生无规分布,并发现有氧化锂生成。V
2
O
5
对过充很敏感,随着嵌入的锂量增加,电荷转移变得不可逆。
晶体型的V 2 O 5 的可逆嵌锂容量比较低,每1mol单元一般可以嵌入不到2mol Li + ,在此范围外便会发生不可逆的相变。但如果制成凝胶,其嵌锂容量会被很大的提高,原因一方面可能是嵌锂位置发生变化,产生热力学上更好的嵌锂位置;另一方面可能是层间距增加,层之间弱的相互作用使得锂更容易嵌入。
层状Li 1+ x V 3 O 8 具有优良的嵌锂能力(每摩尔单元对应3mol以上的锂),且其作为正极时具有比容量高(一般在300mA·h/g)、循环寿命长的优点。其平均电压为2.63V。
其结构是由八面体和三角双锥组成的层状结构,先存的锂离子位于八面体位置,将相邻层牢固地连接起来。过量的锂离子占据层之间四面体的位置,八面体位置与层之间以离子键紧密相连,这种固定效应使其在充放电过程中有一个稳定晶体结构。由于结构稳定以及层之间存在可被锂离子占据的空位,可以允许3个以上的锂离子可逆的在层间嵌入脱出,同时八面体位置上的锂离子从四面体位置向另一个四面体位置跃迁时没有任何障碍,因此具有较高的扩散速率,使得锂在嵌入脱出时具有良好的结构稳定性,从而具有较长的循环寿命。
富锂锰基材料最早由Numata等 [27] 于1997年率先提出,他们提出Li 2 AO 3 ·Li M O 2 固溶体概念,并发现在Li 2 MnO 3 ·LiCoO 2 中,当充电电压达到4.3V时,引入Li 2 MnO 3 可以显著提高材料的循环性能。
单独的Li 2 MnO 3 电化学活性很差,在一般的放电范围内(2.0~4.4V),因为其所有的八面体位置都被占据了,因此不会有锂的嵌入反应;同时,锰离子的高价态很难被氧化,因此也不会发生锂的脱嵌反应。但是当Li 2 MnO 3 与其他层状氧化物材料复合时,得到的复合材料表现出高的可逆容量,与此同时还具有较好的循环性能。因此Li 2 MnO 3 与其他材料复合的体系逐渐变得越来越受青睐,成为正极材料被关注的新宠。
Li 2 MnO 3 为层状单斜结构,可以表示为Li(Li 1/3 Mn 2/3 )MnO 2 ,即可以看作是LiMnO 2 中锰离子层的部分锰被锂取代。由于Li 2 MnO 3 中的锂出现在了过渡金属离子层中,与锰形成了超点阵结构,影响了单斜系的对称性,同时锰离子表现为+4价,这两个因素共同导致了Li 2 MnO 3 电化学性能很差,在传统放电电压范围内呈现出非活性。
富锂锰基材料Li
2
MnO
3
·Li
M
O
2
是由Li
2
MnO
3
和Li
M
O
2
两种材料组成,它们都是α-NaFeO
2
层状结构,属于
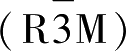 群。不过到目前为止,对于富锂锰基材料的结构还没有达成共识,争论主要集中在过渡金属层中阳离子的排列方式,现阶段有两种主流观点:一种认为Li
2
MnO
3
中的锂离子和锰离子与Li
M
O
2
中的过渡金属实现某种形式的混排,形成固溶体;而另一种认为它们有序排列,形成两相均匀的混合物。
群。不过到目前为止,对于富锂锰基材料的结构还没有达成共识,争论主要集中在过渡金属层中阳离子的排列方式,现阶段有两种主流观点:一种认为Li
2
MnO
3
中的锂离子和锰离子与Li
M
O
2
中的过渡金属实现某种形式的混排,形成固溶体;而另一种认为它们有序排列,形成两相均匀的混合物。
一般认为Li 2 MnO 3 ·Li M O 2 中的Li 2 MnO 3 是惰性的,所以认为它在复合物中仅仅起到稳定材料结构的作用。不过随着研究的进行,人们发现将Li 2 MnO 3 ·Li M O 2 充电至4.5V以上时,会出现一个新的充电平台,且获得高于250mA·h/g的放电容量,且这一容量高于Li M O 2 中过渡金属 M 所能获得的理论容量。因此认为在4.5V以上的高电压下有着新的充放电机制,显然与Li 2 MnO 3 有关。起初人们认为在4.5V的充电平台与四价锰离子被氧化成五价有关,但是原位测试表明,在该电压下锰离子价态并没有发生变化,因此这种新的放电机制不同于传统的层状材料锂的嵌入脱嵌机制解释。
关于Li 2 MnO 3 ·Li M O 2 在4.5V以上新的充放电机制,目前已经提出了几种理论来解释它:Bruce等人 [28-30] 提出的“质子交换机制”,他们在测试富锂锰基首轮充电至不同阶段的元素组成时,发现材料中出现了大量氢元素,并且氢元素与锂脱出量和容量存在一定的对应关系,因此他们认为该处的容量源于锂离子与电解液氧化分解产生的质子发生了交换。Dahn等 [31] 提出“氧流失机制”,这种观点认为在富锂锰基材料的首次充电过程中,锂离子与氧离子同时脱出。Armstrong等人 [32] 认为,首次充电至4.5V以上时,晶格中的氧离子伴随锂离子以Li 2 O的形式脱出,在此过程中,过渡金属离子占据氧离子脱出时的空位,导致脱出的锂离子不能完全回嵌,造成了首次循环的不可逆容量。与此同时过渡金属离子占据锂离子脱出后的空位,导致材料的超晶格结构消失,使得以后的充放电曲线更多地表现为传统层状材料脱嵌锂的机制,4.5V的平台消失。不过到目前为止Li 2 MnO 3 ·Li M O 2 的嵌脱锂的机制还没有被完全了解到。
在Li 2 MnO 3 ·Li M O 2 中,由于放电机制使得Li 2 MnO 3 的首次充放电库伦效率不高,且循环性能很差,而且它是绝缘体,因此倍率性能也不佳。而对于Li M O 2 ,一般来说其循环性能和倍率性能较好,不过容量较低。这两种材料的组合结果使得容量达到了250mA·h/g以上,而且倍率性能和循环性能都得到了一定的改善。
Li 2 MnO 3 ·Li M O 2 目前通常采用共沉淀法,也有其他方法,如溶胶-凝胶法、水热法、高温固相法等工艺来制备Li 2 MnO 3 ·Li M O 2 的,不过因为各自存在的缺点,使得到目前为止,共沉淀法仍然是实验室最理想的制备富锂锰基材料固溶体材料的工艺。
共沉淀法是通过将沉淀剂加入到含有过渡金属离子的溶液中,控制反应条件,使其生成形貌规整,并在原子尺度上组分混合均匀的前驱体,按配比混锂后,再于高温煅烧,最后获得目标产物。共沉淀法通过使几种过渡族金属离子在溶液中充分接触,基本可以达到原子水平混排,使样品形貌形成粒径均匀分布的规则球形,使产物电化学性能稳定。
[1] GUMMOW R J,THACKERAY M M,DA VID W,et al. Structure and electrochemistry of lithium cobalt ox-ide synthesised at 400℃ [J]. Materials Research Bulletin,1992,27(3):327-337.
[2] JULIEN C. Structure,morphology and electrochemistry of doped lithium cobalt oxides [J]. Ionics,2000,6(5-6):451-460.
[3] JULIEN C. Local structure and electrochemistry of lithium cobalt oxides and their doped compounds [J]. Solid State Ionics,2003,157(1-4):57-71.
[4] ZIOLKIEWICZ C. LiCo 1-y M y O 2 positive electrodes for rechargeable lithium batteries:I. Aluminum doped ma-terials [J]. Materials Science and Engineering,2002,95:6-13.
[5] AMDOUNI N,ZARROUK H,F SOULETTE,et al. LiAl y Co 1-y O 2 (0.0≤y≤0.3)intercalation compounds synthesized from the citrate precursors [J]. Materials Chemistry&Physics,2003,80(1):205-214.
[6] JULIEN C,NAZRI G A,ROUGIER A. Electrochemical performances of layered
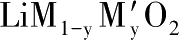 (M=Ni,Co;M′=Mg,Al,B)oxides in lithium batteries [J]. Solid State Ionics,2000,135(1):121-130.
(M=Ni,Co;M′=Mg,Al,B)oxides in lithium batteries [J]. Solid State Ionics,2000,135(1):121-130.
[7] JULIEN C M,MAUGER A,GROULT H,et al. ChemInform Abstract:LiCo 1-y B y O 2 as Cathode Materials for Rechargeable Lithium Batteries [J]. ChemInform,2011,42(12):141-148.
[8] AMDOUNI N,ZARROUK H,JULIEN C,et al.Low temperature synthesis of LiCr 0.3 Co 0.7 O 2 intercalation compounds using citrate,oxalate,succinate,and glycinate precursors [J].British Ceramic Transactions,2013,102(1):27-33.
[9] IRIYAMA Y,KURITA H,YAMADA I,et al. Effects of surface modification by MgO on interfacial reactions of lithium cobalt oxide thin film electrode [J]. Journal of Power Sources,2004,137(1):111-116.
[10] DAHN J R,et al. Thermal stability of Li x CoO 2 ,Li x NiO 2 andλ-MnO 2 and consequences for the safety of Li-ion cells [J]. Solid State Ionics,1994,69:256-270.
[11] ZHANG Z,FOUCHARD D,REA J R. Differential scanning calorimetry material studies:implications for the safety of lithium-ion cells [J]. Journal of Power Sources,1998,70(1):16-20.
[12] CHOI J,MANTHIRAM A. Role of chemical and structural stabilities on the electrochemical properties of layered LiNi 1/3 Mn 1/3 Co 1/3 O 2 cathodes [J]. Journal of the Electrochemical Society,2005,152(9):A1714-A1718.
[13] TARASCON J M,WANG E,SHOKOOHI F K,et al. ChemInform Abstract:The Spinel Phase of LiMn 2 O 4 as a Cathode in Secondary Lithium Cells [J]. Cheminform,1991,22(49):2859.
[14] AYDINOL M K,CEDER G. Firs-principles prediction of insertion potentials in Li-Mnoxides for secondary Li batteries [J]. J Electrochem Soc,1997,144:3832-3835.
[15] DONG H J,SHIN Y J,OH S M. Dissolution of Spinel Oxides and Capacity Losses in 4V Li/Li x Mn 2 O 4 Cells [J]. Journal of the Electrochemical Society,1996,143(7):2204-2211.
[16] JULIEN C M,MAUGER A. Review of 5-V electrodes for Li-ion batteries:status and trends [J]. Ionics,2013,19:951-988.
[17] AMINE K,TUKAMOTO H,YASUDA H,et al. A new three-volt spinel Li 1+x Mn 1.5 Ni 0.5 O 4 for secondary lithium batteries [J]. J Electrochem Soc,1996,143:1607-1613.
[18] OHZUKU T,TAKEDA S,IWANAGA M,et al. Solid-state redox potentials for Li[Me 1/2 Mn 3/2 ]O 4 (Me:3d-transition metal)having spinel-framework structures:a series of 5 volt materials for advanced lithium-ion batteries [J]. J Power Sourc,1999,81-82:90-94.
[19] OKADA S,SAWA S,EGASHIRA M,et al. Cathode properties of phospho-olivine LiMPO 4 ,for lithium secondary batteries [J]. J Power Sources,2001,97-98:430-432.
[20] STRELTSOV V A,BELOKONEVA E L,TSIRELSON V G,et al. Multipole analysis of the electron density in triphylite,LiFePO 4 ,using X-ray diffraction data [J]. Acta Crystallographica,1993,49(2):147-153.
[21] DODD J L,YAZAMI R,FULTZ B. Phase diagram of LixFePO 4 [J]. Eletrehem Solid-Siate Lett 2006,9(3),A151-A155.
[22] XU G,LI F,TAO Z,et al. Monodispersed LiFePO 4 @C core-shell nanostructures for a high power Li-ion battery cathode [J]. Journal of Power Sources,2014,246:696-702.
[23] BYSTROM A,WILHELMI K A,BROTZEN O. Vanadium pentoxide:a compound with five-coordinated va-nadium atoms [J]. Acta Chemica Scandinavica,1950,4:1119-1130.
[24] BACTMANN H G,AHMED F R,BARNES W H,et al. The crysal structure of vanadium pentoxide [J]. Z Kristallogr,1961,115:110-116.
[25] DELMAS C,BRETHES S,MENETRIER M. Cheminform abstract:ω-Li x V 2 O 5 ,a new electrode material for rechargeable lithium batteries [J]. Cheminform,1990,21(35):113-118.
[26] LEGER C,BACH S,SOUDAN P,et al. Structural and electrochemical properties of ω-Li [sub x] V [sub 2]O [sub 5](0.4≤x≤3)as rechargeable cathodic material for lithium batteries [J]. Journal of The Electrochemical Society,2005,152(1):A236.
[27] NUMATA K,SAKAKI C,YAMANAKA S. Synthesis of solid solutions in a system of LiCoO 2 -Li 2 MnO 3 for cathode materials of secondary lithium batteries [J]. Chem Lett,1997,26(8):725-726.
[28] ROBERTSON A D,BRUCE P G. The origin of electrochemical activity in Li 2 MnO 3 [J]. Chem Commun,2002,2790-2791.
[29] ROERSON A D,BRUCE P G. Mechanism of electrochemical activity in Li 2 MnO 3 [J]. Chem Mater,2003,15:1984-1992.
[30] ARMSTRONG A R,ROBERSON A D,BRUCE P G. Overcharging manganese oxides:Extracting lithium beyond Mn 4+ [J]. J Power Sources,2005,146:275-280.
[31] LU Z H,DAHN J R. Understanding the anomalous capacily of Li/Li [Ni x Li (1/3-2x/3) Mn (2/3-x/3) ]O 2 cells using in situ X-ray dffraction and electrochemical studies [J]. J Electrochem Soc,2002,149,A815-A822.
[32] ARMSTRONG A R,HOLZAPFEL M,NOVAK P,et al. Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li[0.2Mn0.6]O 2 [J]. J Am Chem Soc,2006,128:8694-8698.