
下载掌阅APP,畅读海量书库
立即打开



上皮细胞膜渗透性 P ep 可以进一步分解为跨细胞转运的渗透 P trans 和经细胞旁路渗透 P para 两种被动渗透(载体介导的转运将在后面的章节中讨论):

细胞膜是脂质双层,主要由磷脂和胆固醇组成,见图6.4。脂质双层的亲脂性部分成为亲水性分子的渗透屏障。对于可解离型药物,P trans 表达为各组分渗透性之和:


其中,P trans,0 是未解离型药物的固有渗透性,P trans,+ 是电荷为+1的组分的固有渗透性,以此类推。每种组分所占的比例f取决于邻近的上皮细胞膜表面的pH值(微环境的pH在5.5~6.5)和药物的p K a (s),见章节6.1。通常情况下,不带电荷的组分比带电荷的组分更易透膜。因此,根据pH分配理论 ①② 。
① 该关系与辛醇-水分配系数(P oct )和辛醇-水分布系数(D oct )相似,Doct=f 0 P oct ,见7.2节)。
② 最近有报道,离子化的药物分子也可以被动地渗透脂质双分子层(然而,比中性分子慢得多)。

典型的pH-渗透性曲线如图4.7所示。对数图的斜率是1。在此斜率区域,pH或p K a (对数坐标轴)的一个单位差异对应于正常坐标轴上渗透率的10倍变化。因此,可解离型药物在体外渗透实验中采用非生理pH时,使用渗透性值进行生物药剂学建模前,应校正pH值的影响 ① 。当UWL的影响可以忽略不计时,渗透性的水平线与P trans,0 是一致。斜线和水平线的交叉点就是药物的p K a 。但对于UWL限速渗透性的药物,这时水平线低于P trans,0 ,并且交叉点(p K a 变迁)不再是药物的p K a 。
① 体外渗透试验通常使用pH值7.4,但这比微环境中pH值约高1个pH单位。

图4.7 酸性药物的pH-渗透性曲线
游离型药物的固有渗透性P trans,0 可以进一步由药物和脂质双分子层的相互作用推导得出。根据药物分子的性质和膜的组成,计算渗透性的最简单方法是将脂双层作为均匀的有机溶剂膜,并应用Fick’s定律进行计算,见图4.8。细胞膜上的被动渗透是一个扩散过程,透膜的驱动力是膜两侧的浓度梯度差(即Fick’s定律)。如果忽略脂水表面的界面阻力,扩散流量 J 可以表示为:

其中,D mono,m 是药物在细胞膜上的扩散系数,h m 是膜的厚度,C m,0 和C m,h 分别为在膜上位置0和位置h时的药物浓度。C m,0 和C m,h 可以用水和有机溶剂的分配系数K org 表示,和水相中供给侧和接收侧的浓度表示(分别为C w,0 和C w,h )。考虑到漏槽效应,C w,h 将接近0。方程4.32表明渗透性由以下三者决定:分配系数K org (静态参数)、扩散系数D m (动力学参数)和膜的厚度h m ② 。
② 这与Nernst-Brunner方程“固有溶出速率由扩散系数、非搅拌水层UWL的厚度和固体表面的溶解度决定”类似。

图4.8 均匀的膜模型
溶解度-扩散模型可以外推到不均匀的膜模型。渗透系数是渗透阻力的倒数,总的渗透阻力是各个阻力的串联(同Ohm’s定律)。

其中,D m (x)是位置x的局部扩散系数,K org (x)是水和位置x之间的局部分配系数。根据方程4.33,最低的渗透率区域(屏障区域)限制总的渗透率。因此,方程4.33可以简化成方程4.32。K org 是溶质从水(不是从极性基团的表面)到屏障区域的分配系数。目前认为,细胞膜中的扩散系数低于非极性溶剂如十六烷的扩散系数。疏水部分的有序区域(图6.4中的高密度尾部区域)将表现得像是一个柔性聚合物,导致该区域的扩散系数降低。
根据溶解度-扩散模型,膜渗透系数与药物在水和屏障区域之间的分配系数相关。如果能够找到类似于限速屏障的合适有机溶剂,则膜渗透系数可以通过水和有机溶剂的分配系数、扩散系数和屏障的厚度来计算。对于主要由磷脂组成的脂双层来说,建议使用简单的烷烃或烯烃来反映亲水药物的限速渗透屏障。虽然正辛醇是定量构效关系QSAR中最常用的有机溶剂,但它并不很合适。溶解-扩散理论主要用于小分子研究(MW<100),但该理论对类药化合物的适用性未知。
flip-flop机制也作为膜渗透机制被研究。flip-flop机制的概念见图4.9。flip-flop机制用于描述两性大分子或者拟肽分子(如多柔比星)的跨膜运动。这种机制的跨膜运动用两步进行描述:①分子嵌入膜双层结构的小片中;②转移穿过脂质区域(flip-flop)。对于脂肪酸,第一步的嵌入比第二步的flip-flop要快,并且随着链长度的增加,转移穿过脂质区域的速率将减慢。

图4.9 flip-flop机制的膜渗透
正辛醇-水的分配系数通常用作K org 的替代参数。P trans, 0 和P oct 在-2<logP oct <4和0.000 000 1<P trans,0 <0.1cm/s显示广泛且线性的关系 ①② 。用下面的方程粗略估算Caco-2实验的 P trans,0 。
① 注意阶不是10 -6 。最高值0.1cm/s(100 000×10 -6 cm/s)可能看起来很奇怪,实验观察到的最高表观渗透率P app 通常为50×10 -6 cm/s。然而,P app 的上限是由于在标准体外环境下UWL的厚度设置引起的。一旦校正了UWL的影响,P trans,0 可达到0.1cm/s。
② 这并不意味着辛醇和脂质双层对药物渗透具有完全相同的选择性,该关系的标准偏差为一个log单位。


图4.10 非转运体底物在Caco-2细胞中的log P oct 和log P trans,0 的关系

图4.11 有P-gp抑制剂时,Caco-2和MDCK细胞试验中,P-gp底物log P oct 和log P trans,0 的关系

图4.12 采用敲除P-gp的小鼠进行脑灌注实验,log P oct 和血脑屏障固有渗透率 P trans,0,BBB 的关系(或者采用非P-gp底物做本实验)
该方程是用实验P oct 值推导的。P trans,0 是根据Caco-2表观渗透数据计算得到,见章节7.9.5。logP oct 和logP trans,0 之间的关系见图4.10和4.11。图4.11是基于对P-gp底物的被动渗透性的分析得到的,P-gp底物具有较高的分子量,见章节4.9.5。相似的,图4.12显示了被动血-脑屏障BBB的渗透性和logP oct 之间的关系。有趣的是,无论是什么细胞类型(即Caco-2,MDCK和小鼠的BBB),logP oct 和logP trans,0 之间的关系是相似的。中等到高亲脂性且分子量大于500的药物分子(log D oct,pH 6.5 >1.5),将低于中心相关线,且向下偏离。之前的Caco-2的研究中也观测到了类似的偏离,但未排除P-gp的影响。然而,即使剔除P-gp的影响,仍然能观测到这种向下偏离。这一发现表明,小分子和大分子(MW>500)的被动扩散机制是不同的。对于小分子,跨膜渗透可以简单地通过分配-扩散机制描述,而flip-flop机制更适用于大分子药物(MW>500)。方程4.34可以用于MW<500和logD oct,pH 6.5 =2~5的药物,但不适用于MW>500和logD oct,pH6.5 >5的药物。当考虑分子量的影响时,可以获得以下的经验方程(图4.13):


图4.13 非转运体底物 P trans,0 的预测值和观测值
虽然方程4.34和方程4.35只是对游离型药物的固有渗透性P trans,0 的粗略估计,但是这两个方程在药物发现和开发阶段非常有用,特别是对于亲脂性的药物(logD oct,pH 6.5 >2)。由于体外试验存在人为误差,见章节7.9.8,对于高亲脂性的药物(logD oct,pH 6.5 >1.5)存在P app 被低估的固有风险。而实验得到的logD oct,pH 6.5 相对可靠,上限可以高达4。此外,大多数情况下(除了MW>500的药物),当logD oct,pH 6.5 >2时,P eff 主要受非搅拌水层UWL的影响,因此,准确估计P ep 不是必需的。如P trans,0 >0.1cm/s时,不应使用方程4.34和方程4.35,因为这些方程未在此范围内进行验证。P trans,0 有理论上限,该值受药物在细胞质的扩散过程控制。
举例
pH 6.5时,酮洛芬的P trans 可以由logP oct =3.2和p K a =4计算得到 ① ,
① 在转运体文献中经常引用以下误解:p K a 为8.5(碱性)或p K a 4.5(酸性)的可解离药物在中性pH时,有99%是离子化的,不能通过被动扩散透过细胞膜的脂质双层。这时后面通常加上“因此,大部分离子化的药物是通过转运体进行吸收的”。这个误解可能来自忽略了P trans,0 至少可以高达100 000×10 -6 cm/s。

小分子可透过上皮细胞之间的紧密连接。这种紧密连接由细胞黏附分子维持并带有负电荷。阳离子小分子(对于人来说MW<200或<400)更倾向于经细胞旁路途径进行渗透,而大的和(或)带负电荷的分子则不能。经细胞旁路途径的药物渗透模型已用负电荷管道成功搭建。



其中, fz 是用药物的p K a (s)计算得到的每种带电荷组分的分数( z 指的是电荷数目), R ratio 是经细胞旁路渗透的表观孔半径与渗透物的分子半径的比值( R ratio =MW 1/3 /R MW ),经细胞旁路表观孔半径是基于分子量MW(R MW ,人=8.46)的选择性来计算。 A ″是经细胞旁路途径群体的总常数( A ″=3.9×10 -4 ,P Para 单位是cm/s)。Z para 对应经细胞旁路途径的表观电势(肠的电势从-18到80mV)。由于细胞膜带有负电荷,经细胞旁路途径具有阳离子选择性。RK是一个分子筛选函数(Renkin函数)。随着扩散分子半径的增加,RK会降低。虽然细胞旁路途径模型方程是一个近似方程,但它成功地模拟了经细胞旁路途径的贡献,见8.4.4章节。除了分子量MW和电荷数目 z ,底物的亲脂性同样会影响经细胞旁路途径渗透。特定情况下,药物分子形状也同样会影响表观渗透性 P app ,例如PEGs。
经细胞旁路途径的有效范围具有种属差异。化合物在狗中的经细胞旁路途径,比大鼠和人的更易渗漏,见章节13.5.1。Caco-2细胞往往比人体小肠具有更紧密的紧密连接。因此,当我们研究种属差异和体外-体内相关性IVIVC时,应考虑经细胞旁路途径的影响。
图4.14显示了使用方程4.36~4.39计算经细胞旁路渗透P para 而预测吸收百分数Fa%。与被动跨细胞转运渗透相比,经细胞旁路通常被认为是次要途径。但许多亲水的碱性药物(p K a >6.5)会经细胞旁路途径渗透,例如,阿替洛尔(MW=266)、二甲双胍(MW=129)和雷尼替丁(MW=314)。经细胞旁路渗透P para 可以从分子量MW和p K a 估算,且具有合理的准确性,因此计算P para 的收益/投入比高。

图4.14 基于GUT框架,通过经细胞旁路途径估算吸收分数Fa%
(a)在人体;(b)在狗中。
用方程4.35~4.38计算logD oct (pH 6.5)、MW和Fa%之间的关系,见图4.15。在图8.8中,理论计算与实验观测结果非常一致。


图4.15 用方程4.35~4.38计算logD oct (pH 6.5)、MW和Fa%之间的关系
(a)不可解离的药物;(b)酸性药物;(c)碱性药物
图4.16是上皮细胞的示意图。为了更合理地模拟药物在细胞质内的生物过程,细胞质内的药物有效浓度应该定义为游离型药物浓度 f u1 C 1 ① 。f u1 C 1 可能与顶端侧的药物浓度有很大的差异。图4.16显示对过程进行全数值积分,已广泛用于研究肠道细胞中的药代动力学。然而,通过应用稳态近似,净渗透率和f u1 C 1 可以简单地计算,无需数值积分。
① 本节中应用的参数的具体定义请见图4.16。
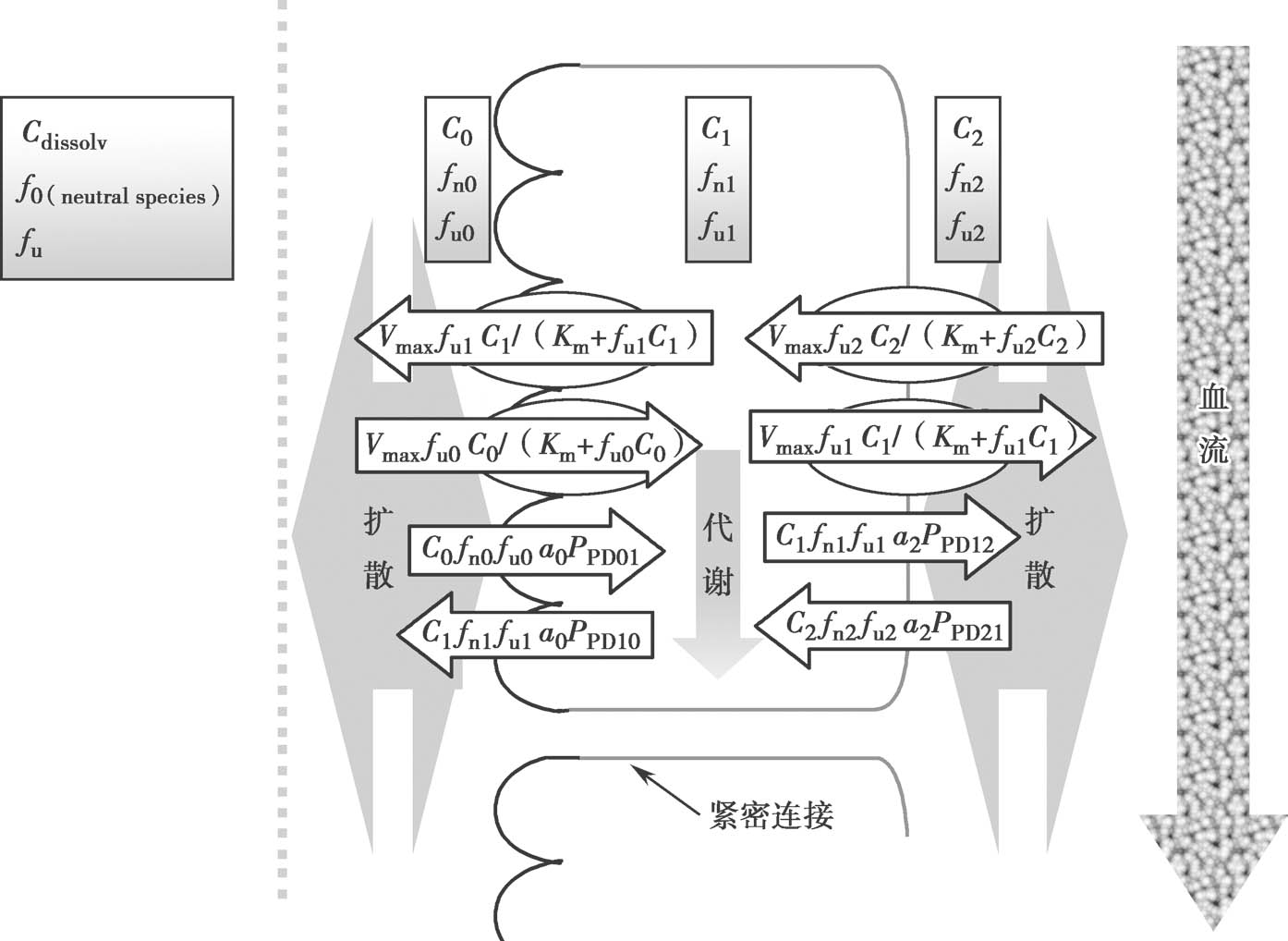
图4.16 肠上皮细胞架构图
在体外细胞膜渗透测定中,在短暂诱导后,接收池中的浓度随时间呈现线性增加,这意味着细胞质中的浓度在诱导后达到了稳定状态。在这个线性范围内,表观渗透率P app 可以用以下方程计算:

其中C donor 是给药池孔中溶解的药物浓度,A well 是孔的表面积,X acceptor 是接收池孔中的药物量。
在大多数情况下,采用稳态近似法是合适的,主要因为细胞质中的液体体积比肠液少得多 ① 。在下一节中,基于稳态近似方法,将讨论细胞模型的理论细节。
① 与给药池浓度变化的时间刻度相比,当细胞溶质中的稳态快速建立时,就可以用稳态近似来解析方程。即使当C dissolv 随时间变化,稳态近似也适用于每个时间点。在稳定状态下,给药池和细胞质中的浓度之比可近似为恒定。
通常,通过载体介导的渗透过程是会达到饱和的,而且底物是有特异性的,该过程也可以被抑制。载体介导的固有渗透率可以表示为:

其中,J max 是最大流量,K m 是米氏常数,C cm 是转运部位的有效浓度。K m 和C CM 定义一致。当C CM <<K m :

J max 与每个酶的表达水平成正比,为了校正体外和体内表达水平的差异,可用下面的方程表示:
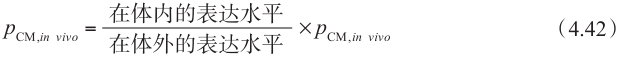
这种最简单的情况下,稳态下细胞质房室的质量平衡方程表达为:
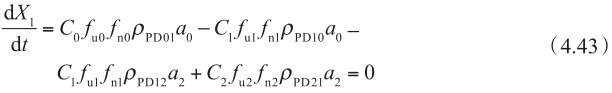
其中,fn=非离解型(不带电荷)药物的百分数;
fu=游离药物的百分数;
p=理想渗透率;
C=每个房室中溶解的药物的总浓度;
a=绝对的表面积;
X=每个房室中的药物量;
PD=被动扩散
0,1和2=图4.16中的房室。
这个方程基于两个假设:
1.只有游离的药物可以渗透过细胞膜(游离组分理论)。
2.只有非解离型的药物可以被动扩散进入膜(pH分配理论)
在稳定状态下,在每个时间点细胞质内的净质量平衡可以近似为零。基底侧C 2 药物浓度远小于顶端侧的药物浓度,可以忽略不计(C 2 =0)。通过重新整理方程4.43,C 1 f u1 可以表示为:
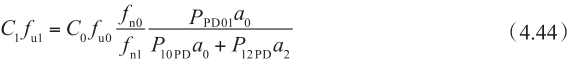
这个方程可以解释为细胞质中游离药物的浓度C 1 f u1 由以下四个因素决定:非解离型药物百分数在顶端侧和细胞室之比值f n0 /f n1 ,固有的被动渗透率,顶端侧游离药物分数f n0 ,表面积 P PD01 a 0 /( P PD10 a 0 + P PD12 a 2 )。f n0 /f n1 可以根据药物的p K a 和顶端侧及细胞质中pH计算得到。因此,稳态下,计算细胞质中游离药物的浓度并不需要知道细胞质游离药物的百分数 ① 。换句话说,游离的非解离型药物的浓度梯度仅仅决定了被动扩散过程。图4.17是依靠浓度梯度扩散穿过肠壁的示意图;图4.18显示了非离解型和解离型药物的浓度曲线。
① 这种情况与PK-PD理论相似,因此靶器官中细胞质内的药物浓度可以通过血浆中药物浓度和血浆游离药物百分数来计算(当不涉及载体介导的转运时)。
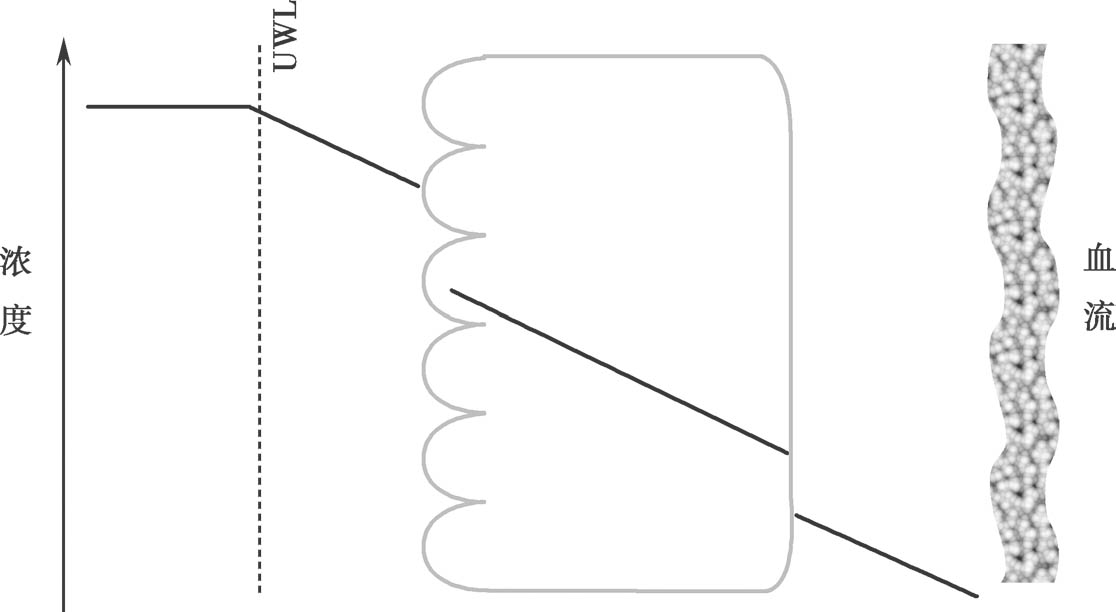
图4.17 跨越上皮细胞膜的浓度梯度示意图(粗线表示穿过肠壁的浓度梯度)
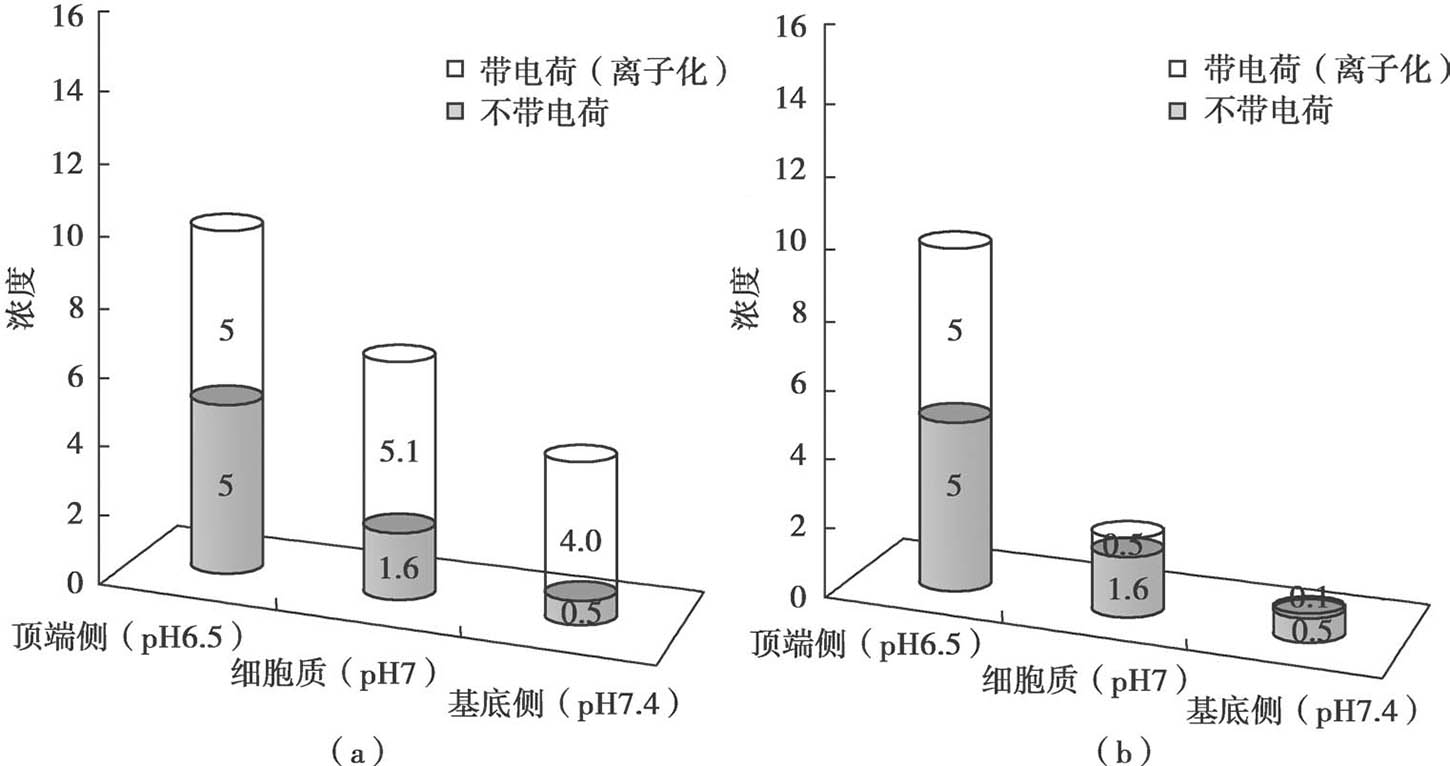
图4.18 上皮细胞中非离解型和带电物质的浓度,游离的非解离型药物的浓度梯度(灰色柱)仅决定被动渗透过程
(a)酸性药物的示意图(b)碱性药物的示意图(两者的p K a 均为6.5)。游离的非解离型药物分子浓度梯度决定被动渗透性
当被动渗透在流入和流出的方向是对称的,且顶端侧和基底侧上的流入和流出是相等的(即 P PD01 = P PD10 = P PD12 = P PD21 = P PD ) ① ,同时上皮细胞的表面积比为1∶3时,则方程4.44变成:
① 这一假设得到以下事实的支持:在不同pH时,顶端侧A到基底侧B和对应B到A的被动渗透率相同。
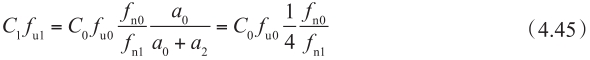
因此,当顶端侧pH与细胞质房室中pH相同(例如pH 7.4),或者药物是不可解离时,细胞质中的游离药物浓度C 1 f u1 是其在顶端侧的药物浓度C 0 f u0 的1/4。当pH不同时(例如在顶端侧微环境为酸性pH 6.0~6.5,细胞质中pH为7.0~7.4),且药物是可解离的时,应考虑非解离型百分数的差异,这对预测肠上皮细胞中药物相互作用DDI特别重要,见14.2章节。
举例
西咪替丁在细胞质/顶端侧中的游离药物浓度比值可以计算如下(p K a =6.9;顶端侧pH=6.5,细胞质pH=7.0,且顶端侧没有胆汁胶束-药物结合):
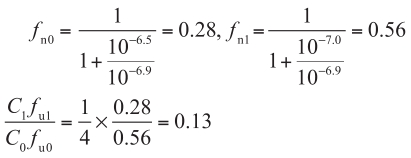
用细胞溶质中的药物浓度计算净渗透率,如下所示。在稳态下,透过顶端膜的流量等于从顶端侧到基底侧的净流量。
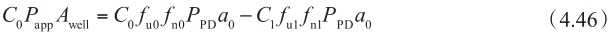
方程的左边是从顶端侧到基底侧的宏观净流量(即表观渗透性P app ),表观渗透率P app 的定义是根据细胞培养孔板的表面积A well 和顶端膜表面的溶解药物浓度C 0 计算。方程的右边是在顶端膜微观净流量。将方程4.44代入方程4.46,得到:
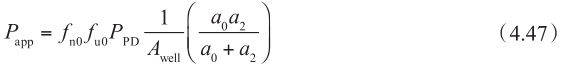
这个方程可以理解为表观渗透性P app 受顶端侧中不带电荷药物百分数f n0 和游离型药物百分数f u0 的影响,但不受细胞质中不带电荷药物百分数f nl 和游离型药物百分数f ul 的影响(该方程中没有f nl 和f ul ) ① 。如图4.18所示,被动渗透的流量仅由非解离型的游离药物的浓度梯度决定 ②③ 。
① 通常认为,pH分配理论应该是不正确的,因为无论顶端侧的pH如何变化,细胞质中pH保持恒定(在pH 7.4)。并且对于酸性药物来说,其基底侧的渗透是限速步骤,即主要渗透屏障(除非顶端侧的被动固有清除率a 0 p 01 远小于基底侧的被动固有清除率a 1 p 12 )。根据方程4.47,细胞质中的pH和游离药物百分数不会影响表观渗透性Papp,因此,无论a 0 p 01 和a 1 p 12 的值如何,pH分配理论都是有效的。文献中的大量实验已证实可解离的化合物都遵循pH分配理论。
② 不应混淆药物浓度和药物百分数。
③ 在原位测定和离体试验中,由于微环境pH的存在,通常pH依赖的药物的渗透不能得到准确的测量,微环境pH可以维持不变,并且几乎不受流体pH的影响
当C 1 f u1 <<K m 时,外排转运体介导的转运可以看作一级动力学过程。从顶端侧A到基底侧B方向,稳态下,细胞质内的质量平衡如下:
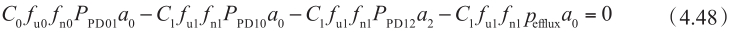
重整该方程,得到:
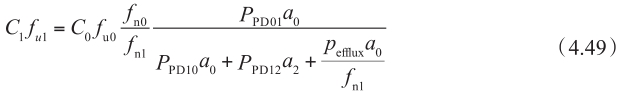
另一方面,根据稳态下表观渗透性的定义和穿过基底膜的流量,得到:
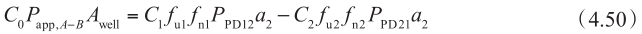
该方程中,稳态下穿过基底膜的流量(方程右边)等于给药侧浓度和表观渗透率的总流量(方程左边)。通常,P app 在C ≈0的时间点和第二项可忽略不计时计算。在方程4.49中代入方程4.50,得到:
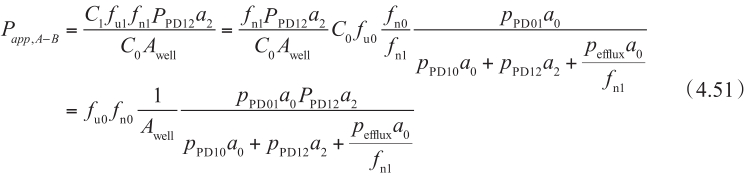
同理,从B侧到A侧方向的方程为:
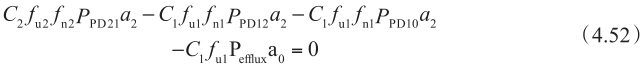
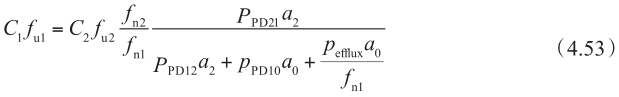
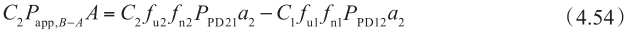
因此,
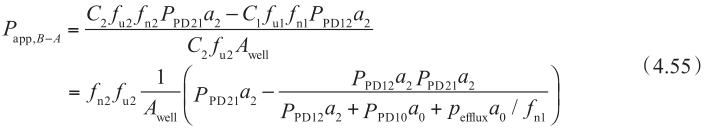
当f n0 =f n2 (不同pH条件下),f u0 =f u2 和P PD01 =P PD10 =P PD12 =P PD21 =P PD ,则外排转运率ER为:

这个方程特别重要,因为它清晰地阐明了外排转运率ER、被动扩散、主动外排转运之间的关系。将方程4.56代入方程4.49中,得到:
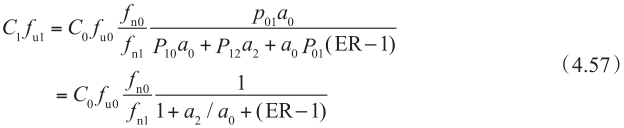
当ER=1,上皮细胞中表面积的比值是1∶3时,该方程代入方程4.44中,得到
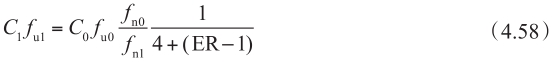
因此,一旦有外排转运率ER数据,就可以估计外排转运体作用下细胞质中的游离药物浓度。
通常,通过抑制实验来估算口服吸收中的外排转运体对其底物的影响。在这种情况下,

这个方程表明,顶端侧外排转运体的抑制引起的AUC的变化比外排转运率ER的变化小得多。例如,当ER=2时,通过抑制顶端侧外排转运体导致的AUC增加是1.25 ① 。方程4.59的另一个优点是可以通过 P app,PD / P app,A-B 估算外排转运率ER。对于很多高脂溶性的P-gp的底物, P app,B-A 超过了体外非搅拌水层 UWL 限制,见章节 7.9.8,而 P app,PD 和 P app,A-B 却没有超过体外UWL限制。这种情况下,方程4.59可以用于计算不受UWL影响的ER。此外,如果没有 P app,PD 数据,可以用 P app,A-B 和 ER 估算 P app,PD (当 P app,B-A << 体外 P UWL 时)。
① 因此,对于有/无外排转运体介导的口服吸收,考虑生物等效性范围(0.8~1.25AUC和 C max ),假设外排转运率ER=2是个很好的标准。
根据方程4.59,

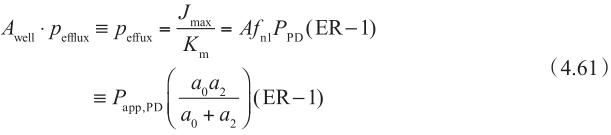
使用方程4.59和方程4.61,可以估算体外和体内的K m /J max 。可以合理假设体内和体外的K m 是相似的。因此,可以通过比较体外和体内J max 值来估算表达水平的区别。这使机制性体外-体内转化IVIVE成为可能。
图4.19中,针对结构不同的药物绘制了体外P efflux 对细胞质中的游离药物百分数f n1 ×被动扩散渗透率P PD 的关系图。P app 数据来自文献。用以下方法估算P efflux :①当P app,PD 、P app,A-B 和P app,B-A 数据已知,且在UWL限制内时,这些数据可用于计算P efflux 和f n1 ×P PD ;②当P app,B-A 超过 UWL限制,而 P app,PD 和P app,A-B 不超过 UWL限制时,用方程4.59计算ER;③当 P app,PD 未知,但P app,A-B ,和P app,B-A 都在UWL限制内时,用方程4.59方程由P app,A-B 和ER计算得到P app,PD 。系统中观测到的最高渗透性的一半被用作UWL限制标准。所有实验在顶端侧和基底侧都为pH 7.4的条件下进行(因此,f n0 =f n1 =f n2 ),且不加入任何增溶剂(f u0 =f u2 =1)。
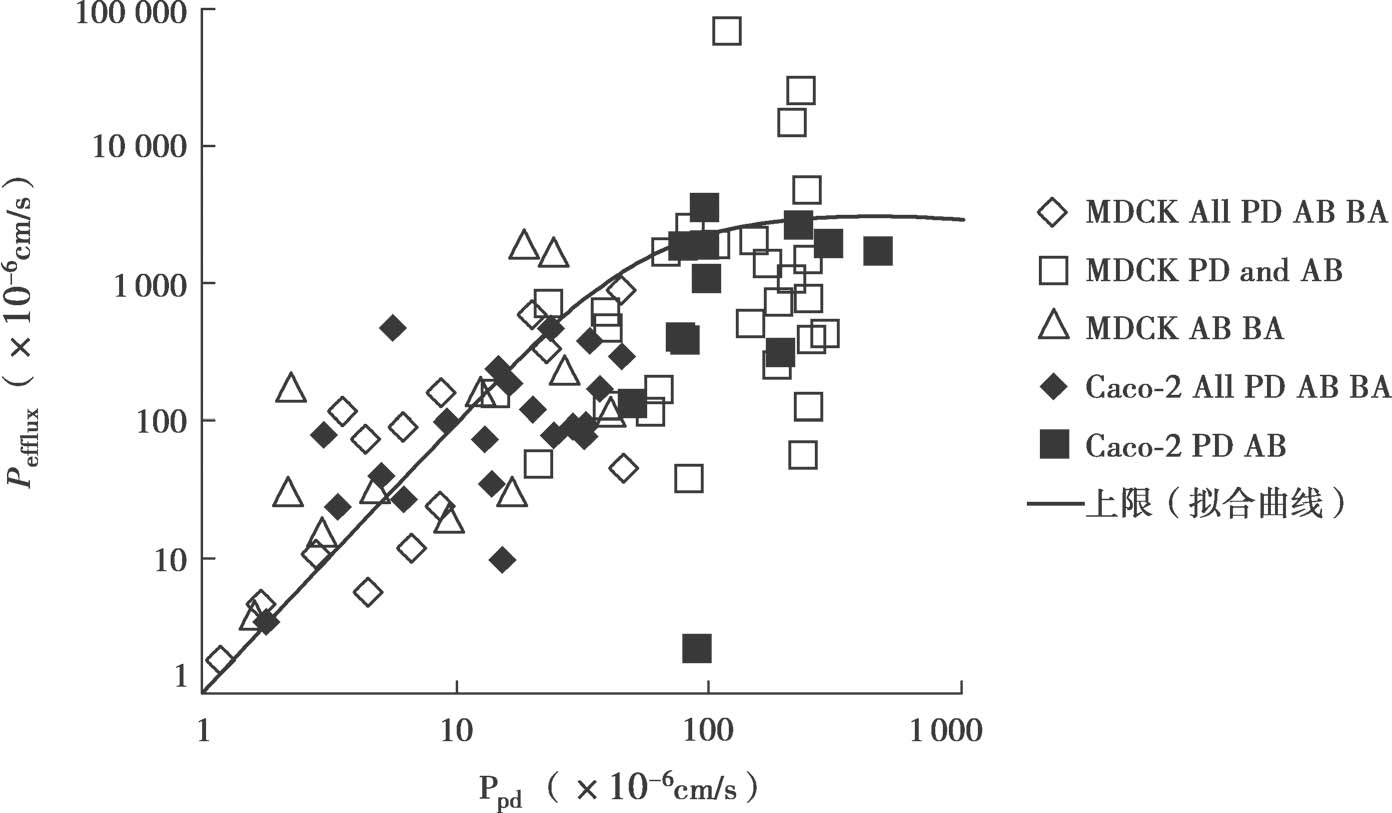
图4.19 p efflux -p PD 关系图
如图4.19所示,在p efflux 和f n1 ×p PD 之间存在相关性。这与P-gp机制非常吻合,即外排转运首先是被动膜扩散步骤,然后是主动跨膜转运步骤,见图4.20。图4.19中的趋势线是:
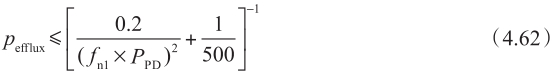
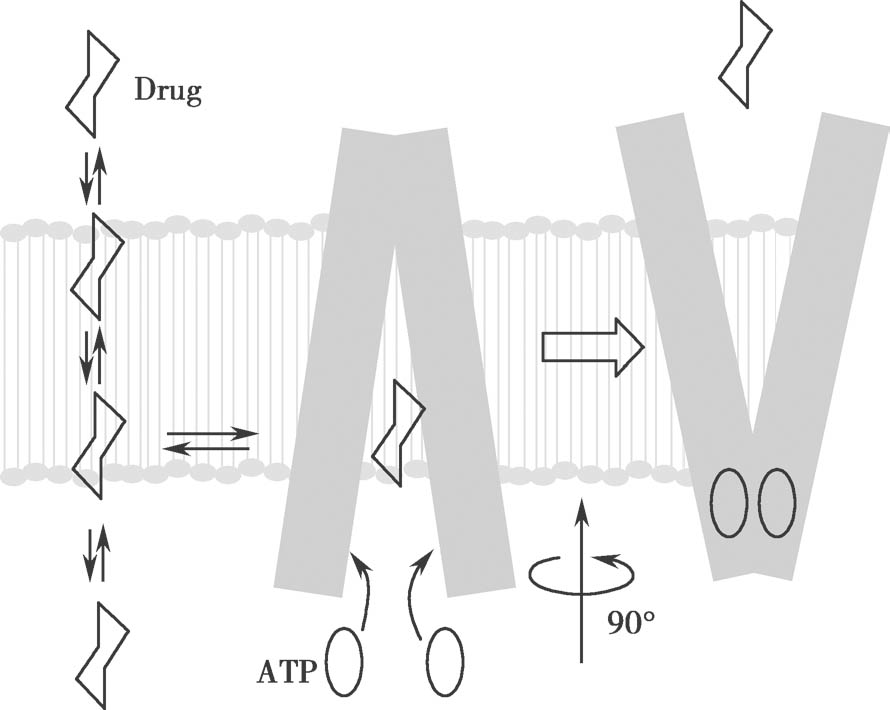
图4.20 P-gp介导的底物转运模型
底物嵌入膜脂双层并通过开放的入口进入内部药物结合口袋,ATP与口袋结合诱导了构象发生大的变化,使药物结合位点产生外排。
其中,P efflux 和f n1 ×P PD 的单位都是10 -6 cm/s。应用这个趋势线,可以计算P-gp对上皮细胞渗透性P ep 、P eff 和Fa%的最大影响。图4.21b显示了抑制P-gp后,不可解离型药物的P ep 、P eff 和Fa%的最大增加值。方程4.35和方程4.62分别用于计算P trans,0 和最大P efflux 。对于中等脂溶性不可解离型药物,抑制/无抑制的P ep 最大比值大约是7,但当被动P ep 高于3×10 -3 cm/s(logD oct 约=2.5)时,P-gp对净P ep 的影响最小。此外,当被动P ep >2×10 -4 cm/s(logD oct 约=1.25)时,净P ep 的最小值大约为50×10 -6 cm/s,且UWL将成为限速步骤,因此即使药物是P-gp的底物,这时P-gp抑制对于总P eff 影响很小,并且可预期口服完全吸收。这些理论推测与实验观测结果非常吻合,当被动P ep 很高时,P-gp对体外ER和体内总吸收几乎没有影响,见章节14.4.2。
此外,对于碱性药物,P-gp对其影响要比不可解离型药物和酸性药物大得多,见图4.22。这是因为顶端侧和细胞质pH值存在差异。当顶端侧pH从7.4变成6.5,且P-gp外排保持不变(细胞质pH一直维持7.4)时,碱性药物的被动流入大约会减小10倍。对于具有中度渗透性的碱性药物,能够观测到P-gp对Fa%的最大抑制作用大约是7倍。
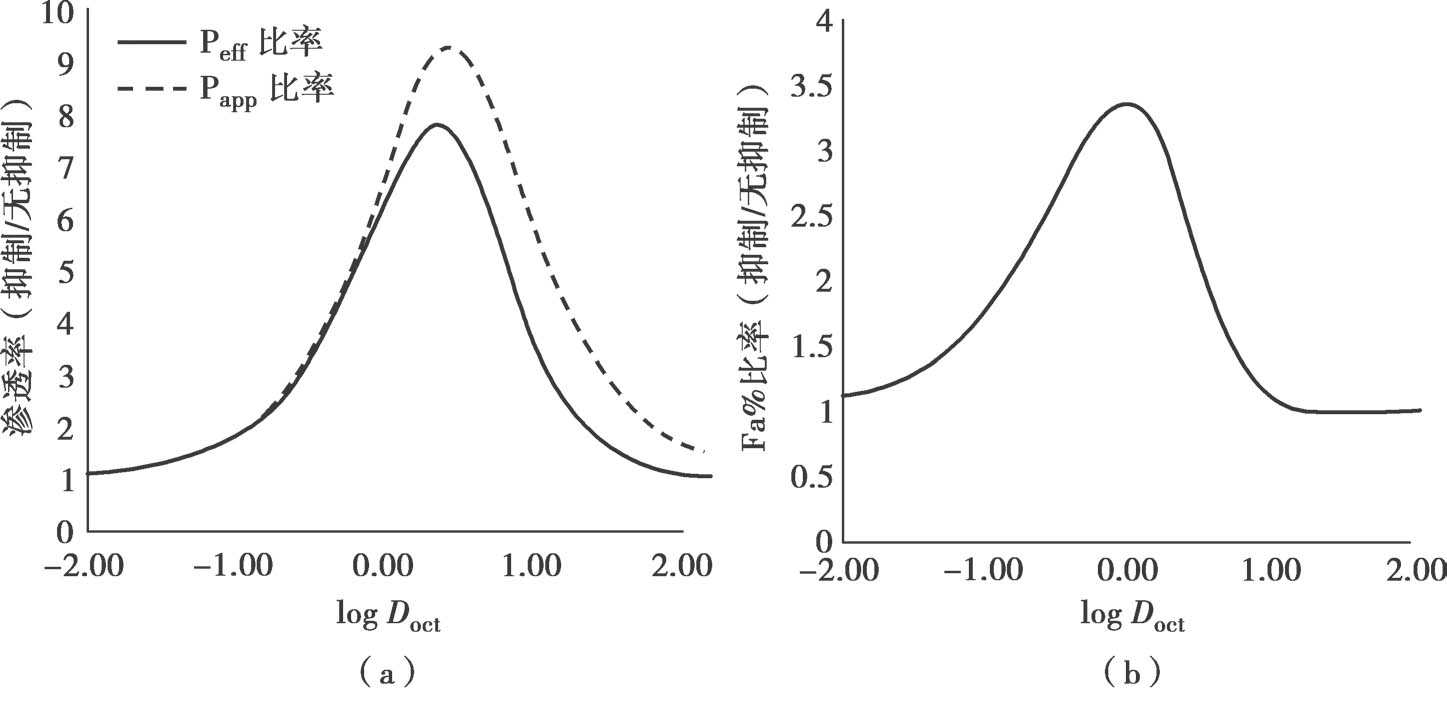
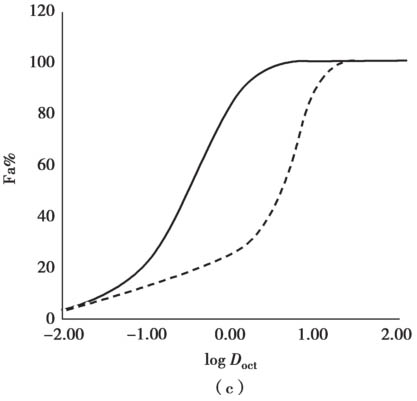
图4.21 P-gp抑制对P eff 和Fa%的最大影响(无溶解度低和溶解速率的限制、无经细胞旁路渗透和胆汁胶束-药物结合,f u =1)
(a)P eff 比率,(b)Fa% 比率,(c)Fa%。PE=3,VE=10,h UWL =0.03cm,D mono =7×10 -6 cm/s,H villi =0.06cm,W channel =0.02cm,W villli =0.05cm,P WC =0.23×10 -5 cm/s
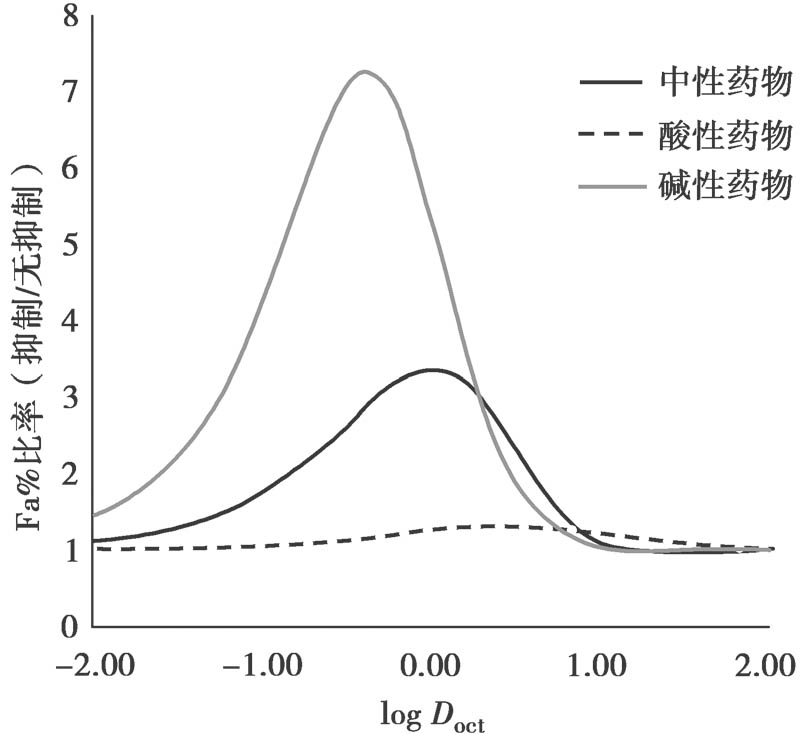
图4.22 P-gp抑制对不可解离药物、酸性药物、碱性药物的Fa%的最大影响
设置酸p K a =4,碱p K a =9。其他条件与图4.21相同。
为了计算外排转运体导致的非线性效应,米氏方程可整合至明确的细胞模型。在这种情况下,细胞房室的质量平衡可写为:
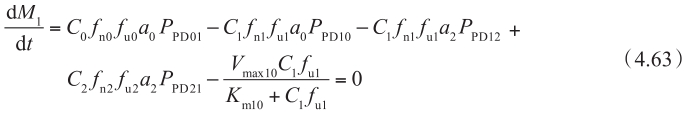
其中K m10 是顶端侧上外排转运体的固有米氏常数,重新整理方程4.63得到如下方程:
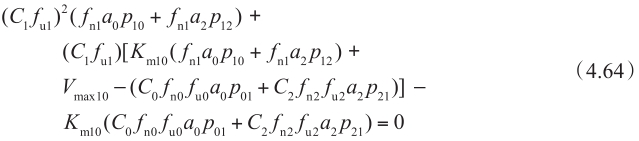
这是C 1 f u1 的二次方程,通过求解方程4.64,可以得到C 1 f u1 :



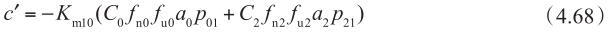
另一方面,根据P app,A-B 定义(方程4.69的左边)和基底侧(方程4.69的右边)的质量转移平衡,可得到:
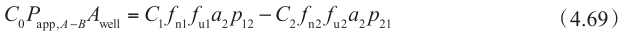
同理,
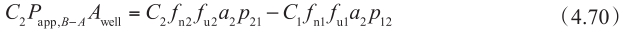
对于从顶端侧到基底侧的渗透,假设在基底侧存在漏槽条件(C 2 =0),可得到:
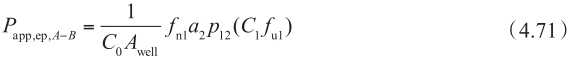
同理,对于 P app,B-A :
这些方程中,C 0 和 C 2 分别是 P app,A-B 和 P app,B-A 的给药侧浓度。将方程 4.65(C 1 f u1 )代入方程 4.71 和方程 4.72 中,可计算 P app,A-B 和 P app,B-A 。
图4.23 在顶端侧膜有外排转运体介导时,浓度-P app 曲线的示意说明
用方程4.62~4.72计算得到浓度-P app 曲线的示意说明见图4.23。很多情况下,对于外排转运体底物,从顶端侧A到基底侧B的渗透和对应B到A的渗透的浓度-P app 曲线是不对称的。这种不对称是由顶端膜和底端膜的被动清除率的差异引起的。图4.24显示了罗丹明123和非索非那定的拟合曲线。此外,表达水平的差异可以改变表观K m 值,因为细胞质浓度是根据P-gp表达水平而变化的。图4.25显示了具有单一的固有K m 值的长春碱的拟合曲线。
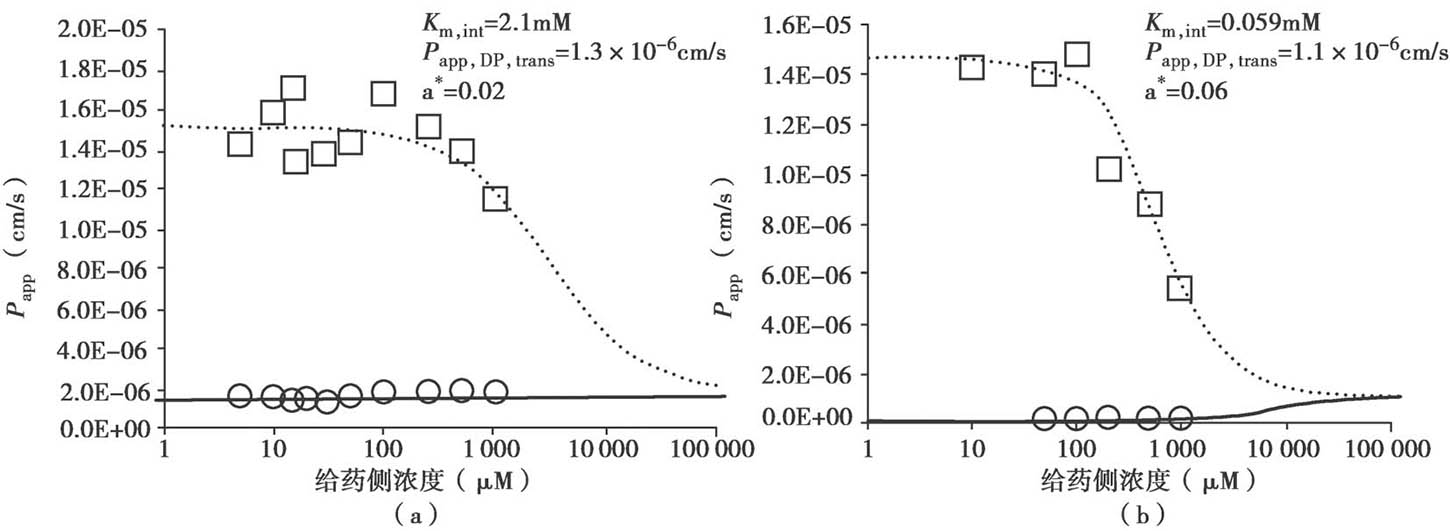
图4.24 罗丹明123和非索非那定实验观测值和模拟的浓度-P app 曲线
(a)罗丹明 123;(b)非索非那定。
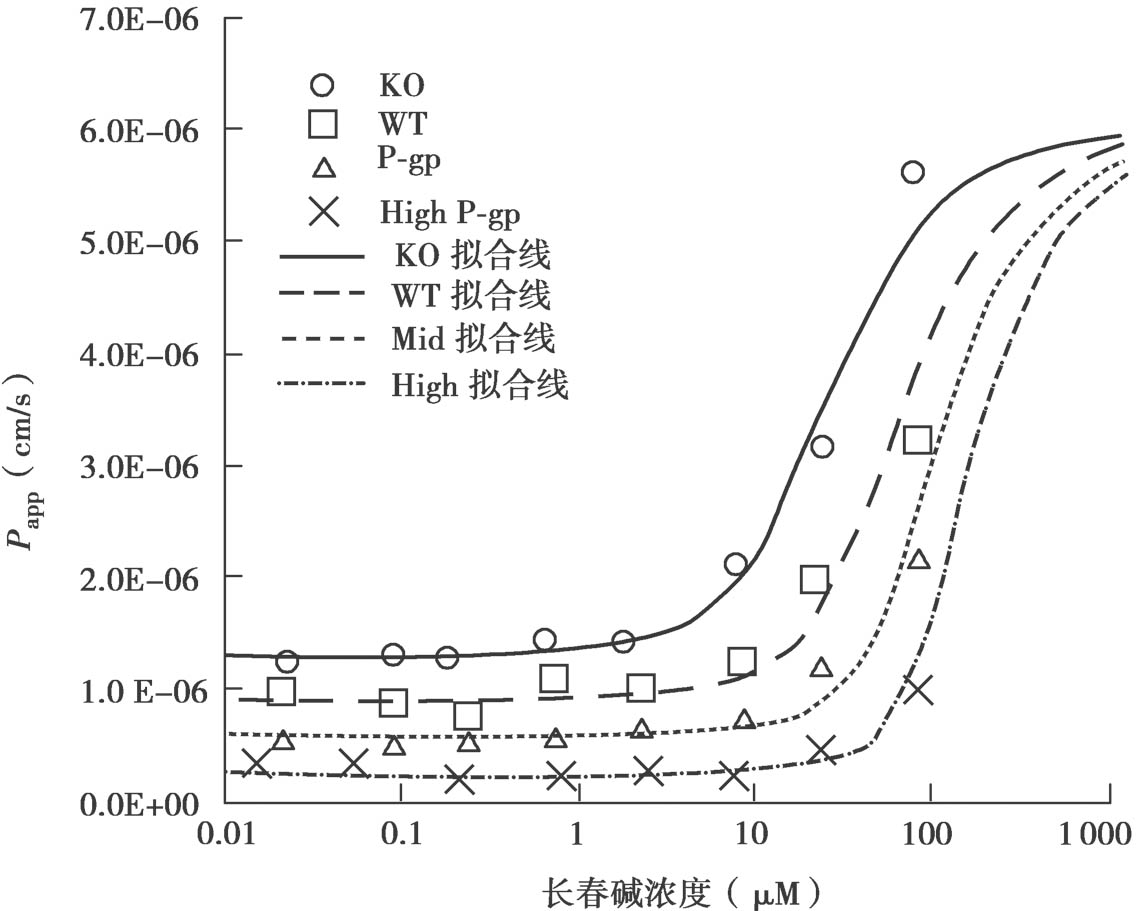
图4.25 长春碱通透性与P-gp表达水平的依赖性
当顶端侧膜涉及摄取转运体参与的膜渗透时,P app,A-B 可以表达为:

当顶端侧膜存在摄取转运体与外排转运体且同时参与转运时,考虑顶端侧上摄取转运体的影响可以通过将等式4.68代替为:
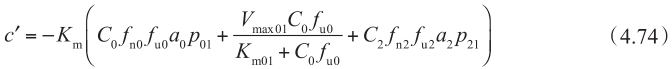
在上述明确的细胞模型的讨论中,非搅拌水层UWL被忽略不计。当考虑UWL的影响时,额外引入以下条件:

其中C 0 是与顶端膜相邻的顶端池的浓度。这个方程意味着在稳态下,透过UWL的流量与透过上皮细胞膜的流量是相同的。
无转运体参与时,重新整理方程4.75,C 0 /C dissolv 可以通过如下方程计算:

当P UWL <P ep 时,由于UWL上存在浓度梯度,上皮细胞膜表面的药物浓度C 0 远比C dissolv 小。此外,对于中性和碱性的药物,f u1 C 1 比f u0 C 0 低约3~10倍(对于酸性药物,f u0 C 0 /f u1 C 1 =2)。因此,为了饱和或抑制细胞质内的代谢酶,给药侧的浓度应该远高于固有K m 和K i 值。当预测药物相互作用DDI时,应把UWL和上皮细胞膜的浓度梯度考虑在内,见章节14.2.2。
当顶端侧有摄取转运体参与时,方程4.73和方程4.75可以解为四元方程(对非线性情况,P ep 是C 0 的函数)。
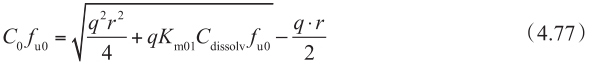

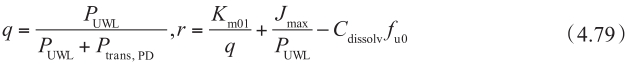
图4.26 显示了顶端侧有摄取转运体参与时,浓度-P app,A-B 的关系。如计算固有K m 时忽略UWL效应,则固有K m 值将被高估(表观K m >固有K m )。
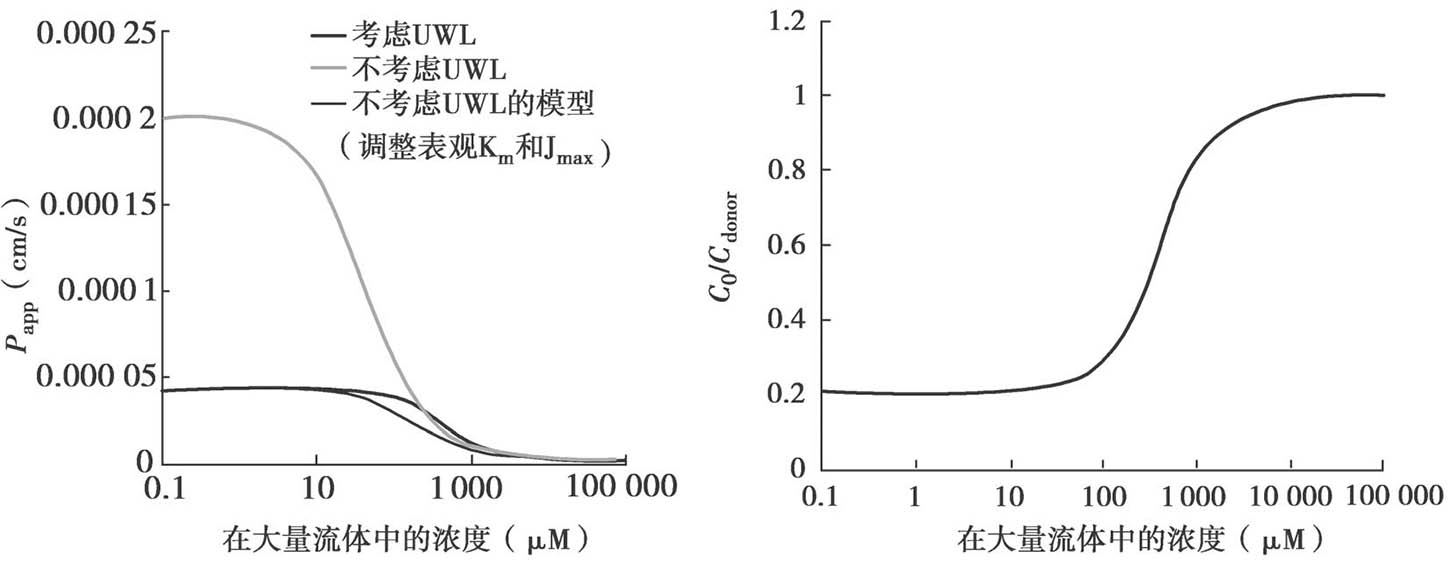
图4.26 在顶端侧有摄取转运体参与时,UWL对底物总渗透性的影响
P active =200×10 −6 cm/s,K m =50μm,P UWL =50×10 −6 cm/s。无被动扩散。
在这种情况下,同时考虑UWL效应和可饱和外排转运体的影响的理论处理是非常复杂的,还没有一个简单开放的解决方案。但以下过程可用于计算稳态下的P app :


在该条件下,可寻找符合方程4.80的C 0 值,例如采用Newton方法或简单方法 ① 。一旦获得C 0 ,P app 可计算为:
① 体内P eff 可以用相同的方法获得,但计算体内Peff时应考虑表面积(皱襞和绒毛)和胆汁胶束-未结合药物百分比。

图4.27显示了非搅拌水层UWL的渗透率 P UWL 是如何影响表观渗透性 P app 的。
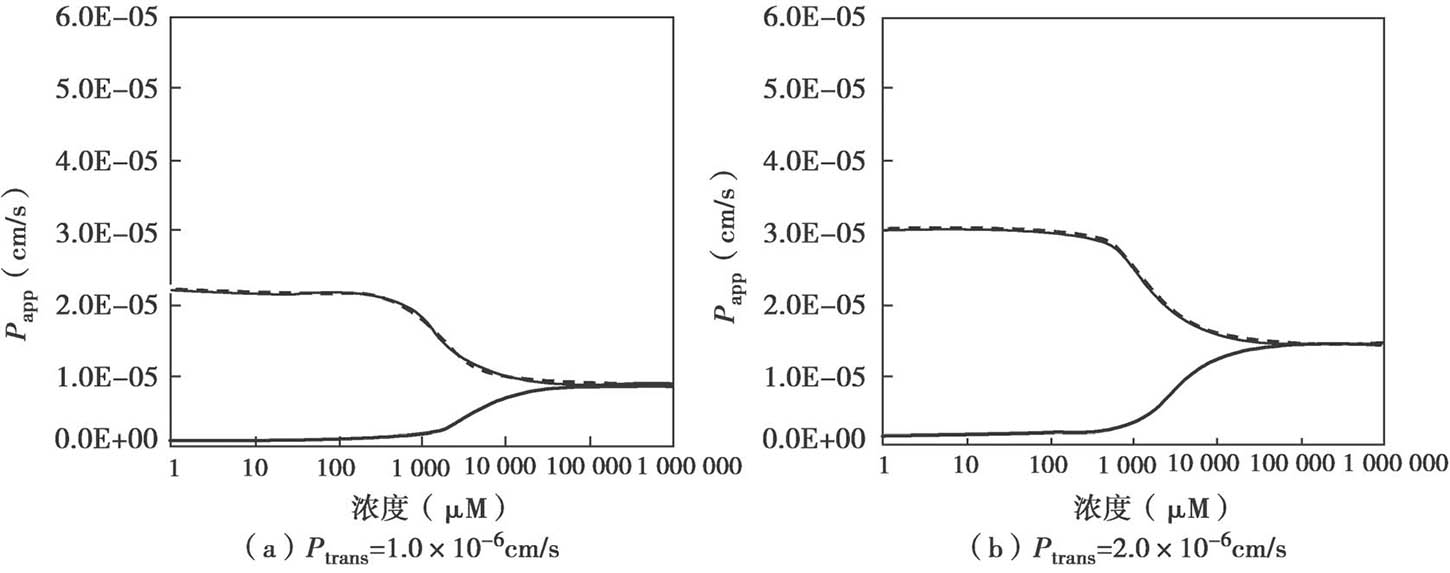
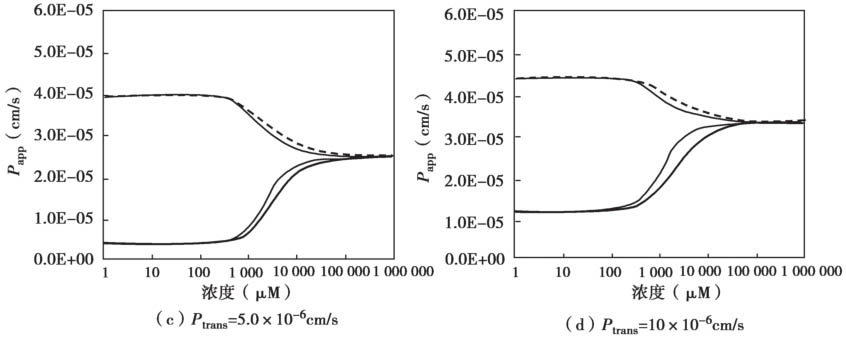
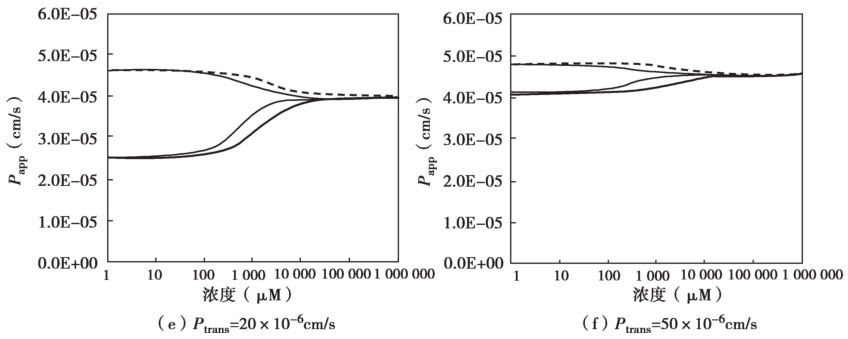
图4.27 非搅拌水层UWL对外排转运的底物总渗透性的影响
主要参数如下:K m =50μM,P UWL =50×10 -6 cm/s;被动扩散P trans 是从 1变化到50×10 -6 cm/s;P efflux 由方程4.62计算得到
肠壁中的代谢是非常显著的,特别是CYP3A4和UGT的底物。已有文献报道了多种预测肠壁中未被代谢的药物分数Fg的方法。本节中,首先讨论文献中报道的两种模型,然后介绍基于解剖学的Fg模型。当细胞质中的游离药物浓度低于CYP3A4的K m 时,可以用这些模型。可以为明确的上皮细胞模型建立微分方程,通过数值求解而进行更高级的模拟。
Yang等人介绍了与充分搅拌模型(well-stirred model)相似的肠血流量Q gut 模式。


其中,Q villi 是绒毛血流量(人的绒毛血流量是18L/h)。在肠血流量Q gut 模型中,PS perm 定义为肠膜有效渗透性,PS perm =肠的光滑表面积(0.66m 2 )×P eff ,P eff 可由P app 体外试验(MDCK和Caco-2)数据通过简单的线性回归估算。据报道,直接设定f u1 =1可得到最好的预测结果,而不是采用血浆游离百分数f up 作为f u1 的替代值(假设f u1 =f up )。但报道中未给出明确的解释,稍后会讨论这种差异的一个可能原因。图4.29显示了用肠血流量Q gut 模型对Fg的预测,假设f u1 =1。
Kato提出了一个简单方程,根据肝的固有清除率估算Fg :

该方程对于具有高渗透性的CYP3A底物是有效的。
本节中,为了理解肠血流量Q gut 模型和其他模型的背景知识,将讲解理论方程是如何从上皮细胞和肠绒毛的解剖结构中推导出来的。由于从解剖角度推导Q gut 模型并未在原始论文中披露,本节将试图在原始研究者研究的基础上,重现方程的推导过程。
如图4.28所示,Fg基本上由药物的代谢率(k met ×C 1 ×V 1 ;V 1 是细胞中的液体体积)与药物穿过基底侧膜的逃逸速率(k esc ×C 1 ×V 1 )之比决定。当逃逸速率变快时,Fg就会变大,并趋近于1。这是用于所有Fg模型中的主要理念,Fg是逃逸速率占总速率的比值。


图4.28 Fg、被代谢的药物流量和逃逸的药物流量之间关系的示意图
(a)低 Fg;(b)高 Fg
药物代谢和基底膜渗透均由游离药物浓度驱动。因此,在这个方程中,f u1 项在分子和分母中被抵消了。药物从细胞质的逃逸是一系列连续过程,第一步是在基底膜的渗透,第二步是从基底膜扩散进入毛细血管,第三步是通过血流转运到全身的过程。这三个步骤任一过程都可以是限速步骤。
基底膜渗透限速
当基底膜的渗透清除率比其他两个过程慢得多时,

其中,PS baso,int 是游离药物分子在基底膜的渗透清除率。对于被动扩散,可用PS baso,int =a 2 P PD12 表示。在这种情况下,不需要对f u1 进行校正(即f u1 =1),这与Yang等人的发现非常吻合。
血流量限速
当基底膜渗透和穿越上皮下区域扩散无限快时,那么逃逸速率主要受到血流量的限制。逃逸速率等于药物被血流量消除的速率。因此,

等式左边是逃逸速率,右边是血流量的消除速率。在这种限制条件下,细胞质中游离药物浓度和血浆中的游离药物浓度相同,因为细胞质和血浆之间的平衡迅速建立。

其中f up 是血浆中的药物游离百分数。因此,将方程4.89代入方程4.88中,CL esc,int 变成:

基底膜渗透和血流量共同限速
比较这两种情况,一个通用的方程可以用于表示这两种情况,

当 a 2 p 12 >>Q villi /f up 时,CL esc,int =Q villi /f up ,但当 PS baso,int <<Q villi /f up 时,CL esc,int =PS baso,int 。为了推导出Q gut 模型,可以根据C 1 计算基底膜的表观渗透清除率PS perm :

如假设f u1 =f up ,并重新整理方程4.92,将其代入方程4.86,就可得到等同于Q gut 模型的方程:


然而,在肠血流量Q gut 模型中PS perm 的定义是比较含糊的。Yang等人报道了f u1 =1可以得到最好的预测,而假设f u1 =f up 却得到较差的预测。这与基底膜渗透限速的情况是一致的,但是与血流量限速的情况是不同的。
在肠血流量Q gut 模型中,上皮细胞下区域的扩散被忽略。如上皮细胞下区域的扩散是限速步骤,根据与血流量限速的情况相似进行讨论,



其中CL subepithelial 是上皮细胞下区域的渗透清除率,C subepithelial 和f subepithelial 分别是上皮细胞下区域的药物浓度和药物游离百分数。在这种情况下,假定上皮细胞下区域的药物流量与血浆相同(f subepithelial =f up )。则上皮细胞下区域的扩散清除率为:

其中,h subepithelial 是上皮细胞下区域的厚度。这个清除率是基于上皮细胞下区域的总浓度和绒毛血管中的表面积(A blood vessel )计算的。当f up <0.05时,CL subepithelial 变得相对恒定,主要由白蛋白-药物结合分子的扩散系数决定(D albumin =6.6×10 -7 cm 2 /s)。
当把这三种限速的情况合并到综合方程中,它就变成了:

这在本书中将其称为解剖学Fg模型。下一步是评估哪个过程倾向于成为限速步骤,上皮细胞下区域扩散还是血流量。考虑到绒毛的结构,见图6.2,人体中的h subepithelial 可能接近50μm,A blood vessel 大约为 100cm 2 。在这种情况下,CL subepithelial 可能小于 Q villi 。当f u1 <0.05时,CL subepithelial 近似为常数,约2~4ml/min/kg(h subepithelial =50μm和A blood vessel =88.4cm 2 (12.6cm 2 /kg))。CL subepithelial 值偶尔会接近Q villi ,但是它一定比Q villi /f ul 小很多。这可能就是在Q get 模型中,为什么f u1 =1时能够得出较好预测的原因(因为PS baso 限制药物在验证数据集里很少)。
在Q gut 模型中的另一个矛盾之处在于,它表明随着Fg增加,将出现正向的食物效应(吸收增大)。Q gut 模型表明Fg会受Q villi 变化的影响。食物的摄取会使肠血流量增加100%,见图6.23。如Q gut 模型是正确的,那么对于有高Fg的亲脂性药物,预计会产生正向食物效应。但这与实验观测的现象相矛盾,见章节12.2.2.2。Q gut 模型的这两个矛盾可以用解剖学Fg模型来解决。因为当f up <<0.05时,CL subepithelial 接近Q villi ,Q gut 模型(设置f u1 =1)和解剖学Fg模型能得到相似Fg值。此外,因为CYP3A4底物往往具有低的f up 和高的渗透性,这时解剖学Fg模型可以简化成Kato Fg模型,该模型用了固定的CL esc,int 值(人的数值为5.7ml/min/kg)。
Q gut 模型和解剖学Fg模型的主要区别是计算CL esc,int 的方法。直接用实验得到的CL esc,int 比较 Q gut 模型和解剖学 Fg 模型,Fg 可用 CL esc,int 表示:

CL met,int 从人肠细胞微粒体体外实验测得到。这些数据是从多篇文献中收集的,统一用咪达唑仑的CL met,int (6.2ml/min/kg)进行校正,见表4.1。图4.29显示了用Q gut 模型和解剖学Fg模型预测CL esc,int ,的比较。虽然f up =f u1 被认为在理论上更合适,但如使用f up =f u1 ,Q gut 模型很大程度上会高估CL esc,int ,而假设f u1 =1能得到很好的预测,这与Yang等人先前的研究结果非常吻合。通过Q gut 模型(f u1 =1),很多脂溶性的药物CL esc,int ,变成一个恒定值(=Q villi ),以至于更接近Kato的简单模型。解剖学Fg模型可以表现CL esc,int 和f up 之间的依赖关系,见图4.29c。
最近,CYP3A4和P-gp之间的协同作用已被提出,这些酶具有重叠的底物。然而,“协同作用”还没有明确的定义,对实验数据的理解也存在争议。最近,Fan和同事用三房室模型进行了全数值模拟以解决争议。在本节中,将使用稳态解决方案讨论这一点。通过全数值模拟和稳态分析解决方案获得的结果本质上是相同的,但后者将更易于解释并适用于此类书籍。
表4.1 临床实测的Fg,CL int,met 和预测的Fg,CL int,met

续表



图4.29 比较Q gut 和解剖学模型对CL esc,int (a)和Fg(b)的预测性比较及CL esc,int 和f up 之间的关系(c)
Q villi =4.2ml/min/kg,h subepithelial =0.000 5cm,A vlli =126cm 2 /kg,A vein =1.43cm 2 /kg,D albumin =0.66×10 -6 cm 2 /s,PS baso,int =f n1 ×P trans,0 ×(a 0 a 2 /(a 0 +a 2 ))/A well ×a 2 ×A valli 。P trans,0 通过将实验获得数据 logP oct 和分子量 MW 代入方程 4.35 计算得到
如上所述,Fg由药物的代谢清除率和逃逸清除率的比值决定的,见图4.28。因此,如果这两个过程的浓度都是线性的,那这时,顶端侧膜的P-gp将不影响Fg,并且P-gp和CYP3A4之间的协同作用也不会影响Fg。但代谢清除达到饱和后,情况就不同了。
以外排转运体相似的方程开始,在稳态时细胞质中的质量平衡可以描述为:

将顶端侧膜外排转运(一级动力学)加入方程,并将可饱和过程修改为代谢清除过程(CL met,int = V max,met /(C 1 f u1 +K m,met ))。这个方程可以用 C 1 f n1 作为二次方程求解,代入方程 4.69,得到:

图4.30显示了细胞质中药物浓度依赖于逃逸的药物流量和被代谢的药物流量。当顶端侧上的外排转运体被抑制时,细胞质中的药物浓度将增加,且逃逸的药物流量也成比例地增加(因为它遵循一级动力学) ① 。当f u1 C 0 <<K m,met 时,被代谢的药物流量也成比例增加(因此Fg保持不变)。
① 不要与清除率混淆。

图4.30 逃逸的药物流量、被代谢的药物流量、细胞质中的药物浓度的关系图
但对于非线性的情况(f ul C 0 ≈ K m,met ,或 f ul C 0 >K m,met ),被代谢的药物流量不按正比增加。因此,Fg是增加的。
即使外排转运体导致Fa的降低不显著,但仍可观测到外排转运体对F g 的影响。在图4.31中,当P ep >10×10 -6 cm/s时,在浓度范围内Fa%>99%。


图4.31 F g 、P ep (a)、细胞质浓度(b)、被代谢的药物流量(c)与顶端侧的药物浓度的依赖关系
P trans = 5×10 -5 cm / s,p efflux = p PD ×10,K m,met =1μM
目前主要使用表达CYP3A4的Caco-2细胞研究P-gp对于代谢速率的影响。当P-gp被抑制时, F g 会増加。同时,生成的代谢产物量略有増加(因为细胞质的药物浓度増加会引起被代谢的药物流量略有增加)。这一实验结果与理论非常吻合。
理论上认为,即使假定P-gp外排是线性的,P ep 也会表现出浓度依赖性。这是由于药物代谢会引起细胞质的游离药物浓度的改变。当代谢清除饱和时,细胞质中游离药物浓度会增加,因此,顶端膜的浓度梯度会降低,导致P ep 降低(P ep 是对应顶端侧浓度降低率的宏观渗透性,而不是每个膜的固有渗透率,p 01 和p 02 是常数) ① 。
① 在体外测试中,假定接收池中药物的出现量与顶端侧减少的药物量是相等的,P app 通常从接收池中的药物出现量计算得到。但是细胞质中发生代谢降解时,这就无效了。如细胞质中发生大量的代谢,那即使顶端侧渗透很快,接收池中出现的药物量也可能很小。Fa对应于细胞质中出现的药物量(代谢前)并等于从细胞质消失的药物量。