
下载掌阅APP,畅读海量书库
立即打开



药物动力学- 药效动力学(pharmacokinetics-pharmacodynamics,PKPD)综合研究机体用药后药物在体内的经时过程与药效从产生到加强、继而下降、直至消失的经时过程之间的关系。它将2 种不同形式的过程复合为统一体,建立剂量、浓度与效应之间的关系,描述和预测某一剂量药物的效应-时间过程的关系,通过血药浓度-时间-效应三者数据的测定,拟合出血药浓度及其效应经时过程的曲线,推导出产生效应部位的药物浓度,定量地反映其与效应的关系。
进行PK-PD 研究的先决条件是:除血药浓度的经时过程可以被准确测定外,药效强度的指标也应具有明显的可量度以供经时性监测,即可使用仪器直接来测量药效强度,如直接测量瞳孔大小、眼压、血压、体温、尿量,甚至智商分值等。当药物的药效不能量化测定时,就无法用PK-PD 进行研究。
PK-PD 研究一般按以下程序进行:
1.实验中同时测定用药后的血药浓度经时数据及药效变化的经时数据。
2.根据血药浓度数据进行药动学解析,求算药动学参数。
3.判断药效作用的部位(药效室)及作用方式。
4.明确药效室中的药量(浓度)与效应之间的量效转换关系,求出量效转换公式。
5.将量效转换关系与药动学模型中的药效室联系起来,构建PK-PD 模型。
多数药物随给药剂量的增加,体内的血药浓度按比例升高,其药理效应也相应增强。然而血药浓度与药理效应之间并不是一个简单的比例关系,随意加大剂量,往往并不能获得预期效果。药理效应的大小不仅与血药浓度有关,而且更与效应部位浓度有直接关系。因此,研究血药浓度和药理效应之间的关系尤为重要。
有些药物的血药浓度和药理效应呈一定的线性关系,即随着血药浓度的增加,药效也逐渐增强,最终达到一个最大值(图4-11);以药效对血药浓度的对数作图,可得到一个S 形曲线或者抛物线(图4-12)。许多药物的治疗浓度范围通常落在其药理效应强度达到20%~80%所对应的血药浓度范围内,这时的药理效应强度和血药浓度的对数呈现良好的线性关系。
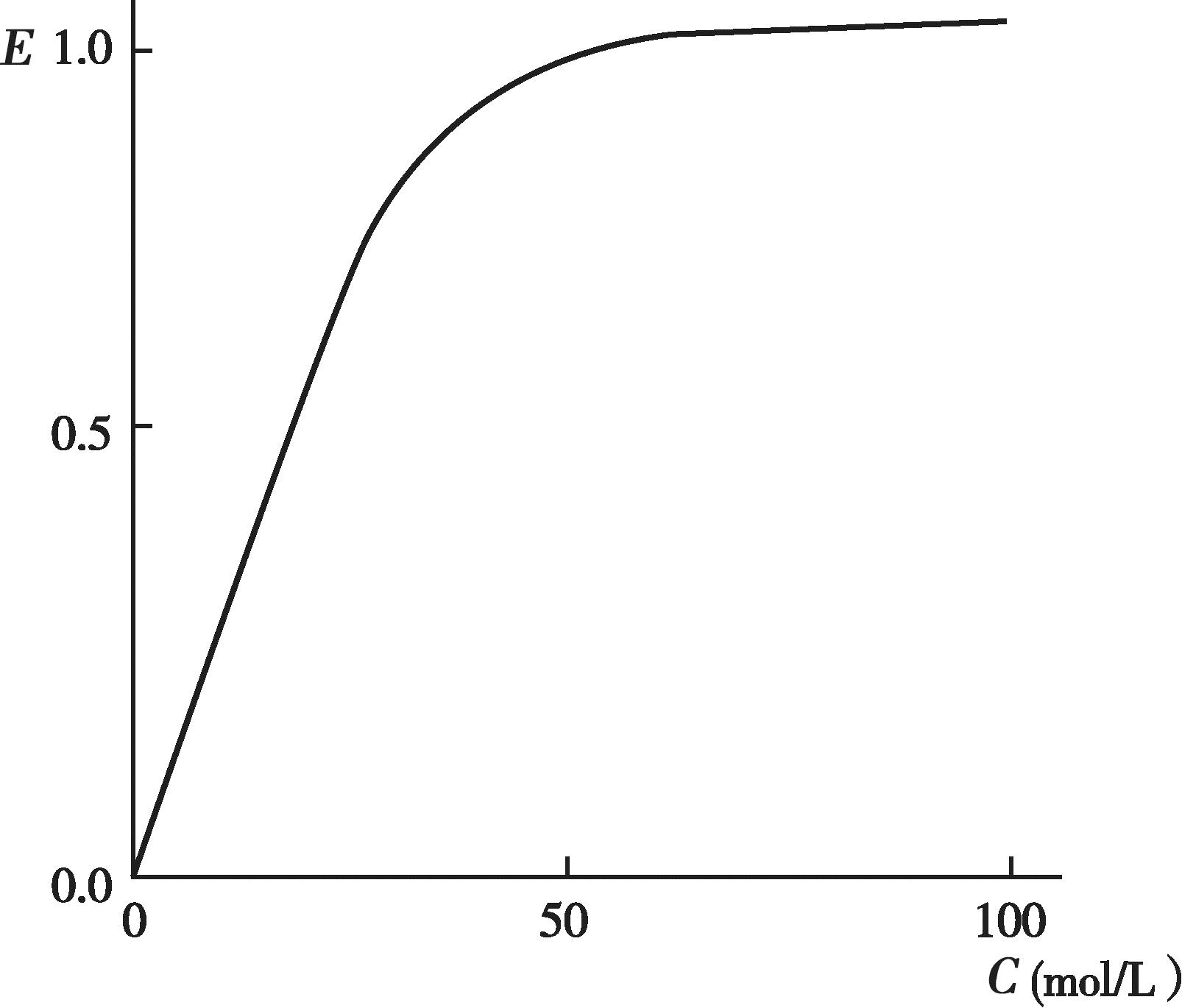
图4-11 药效和血药浓度曲线图
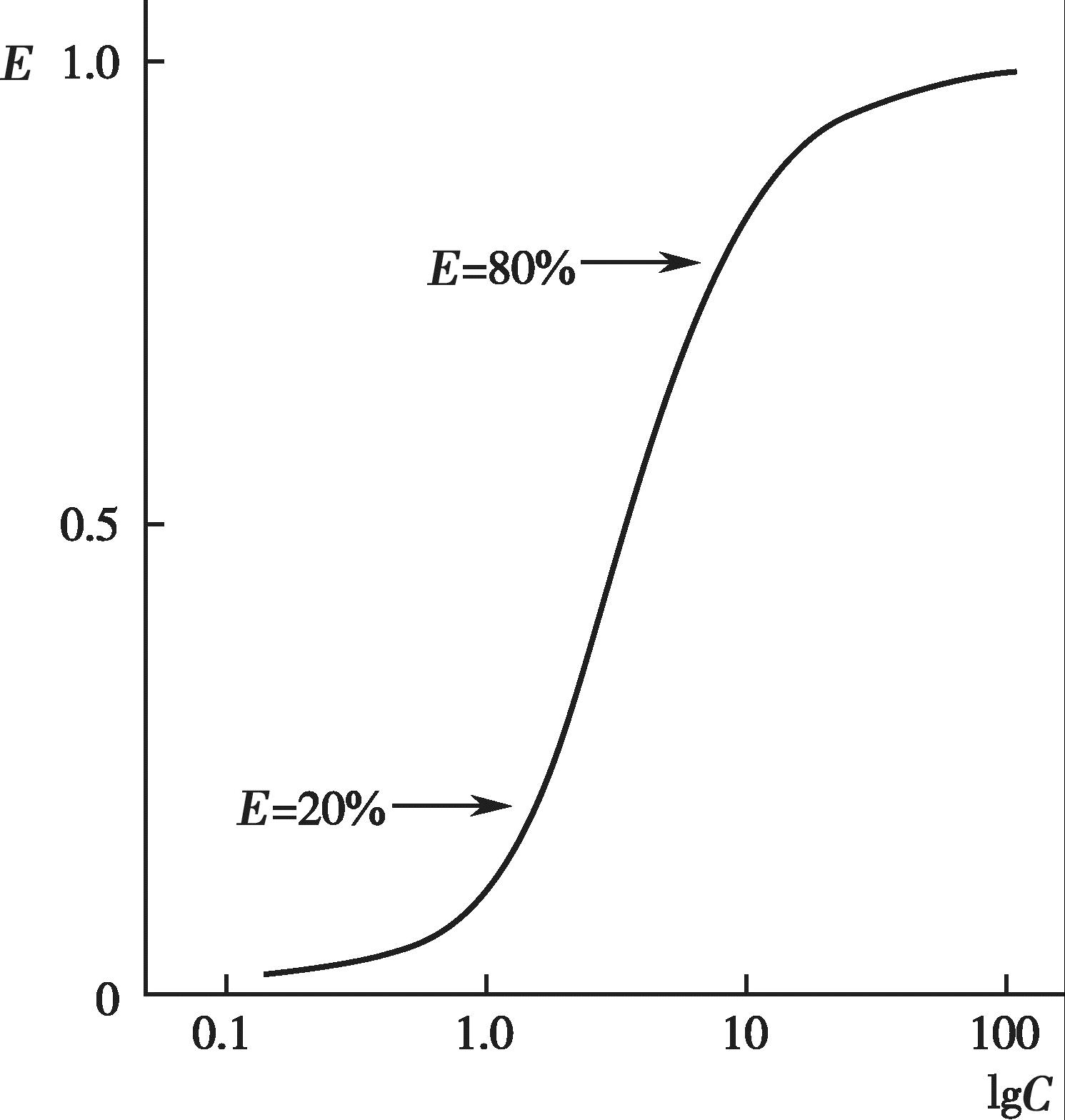
图4-12 药效和血药浓度半对数曲线
有些药物的血药浓度和药理效应不是呈简单的线性关系,当药理效应在血药浓度很低时其强度增加不明显,而当血药浓度达到一定值后会出现明显的增强,但该显著增强的趋势随着血药浓度的不断增加又逐渐减小,并趋近于最大值,这就是所谓的S 形曲线。许多药物在体内的药效-剂量曲线都符合S 形曲线的特点。
有些药物的血药浓度和药理效应不平行,表现出一种间接的关系。这种间接相关主要有以下几种:
(1)血药浓度-效应的逆时针滞后曲线:
某些药物的血药浓度-效应曲线(counter clockwise hysteresis)呈现出明显的逆时针滞后环,如图4-13 所示。图4-13 中的箭头表示时间的走向,从曲线可以看出,给药后每一时间点上的浓度和效应不是严格的一一对应关系,效应峰值明显滞后于血药浓度峰值,这表明效应室不在中央室,而在周边室。即初始血药浓度很高时,其药理效应并不强;当血药浓度进一步分布到周边室后,才逐渐产生药理效应,至中央室和周边室血药浓度达平衡时,药理效应达最大,随后药理效应才随着血药浓度的衰减而减少,因而出现效应滞后于血药浓度的现象。如盐酸普萘洛尔、麦角胺等药物即属此类。
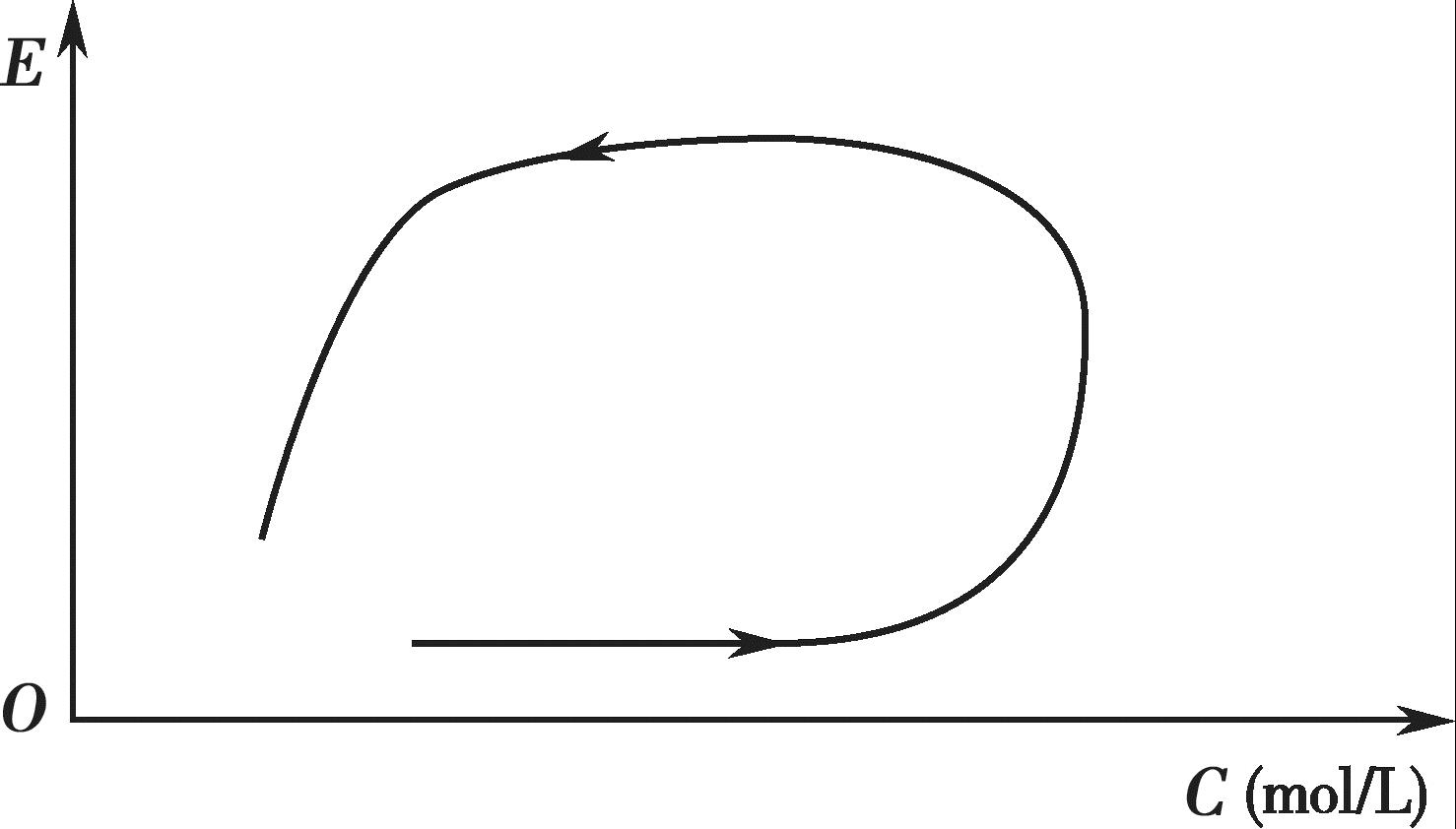
图4-13 血药浓度-效应的逆时针滞后曲线
(2)血药浓度-效应的顺时针曲线:
某些药物的血药浓度- 效应曲线(clockwise hysteresis)呈现出明显的顺时针环,如图4-14所示。从图4-14 中可以看出,与血药浓度上升期相比,下降期内同样的血药浓度所对应的效应明显减弱,这表明该药物在体内可能出现了快速耐受性。
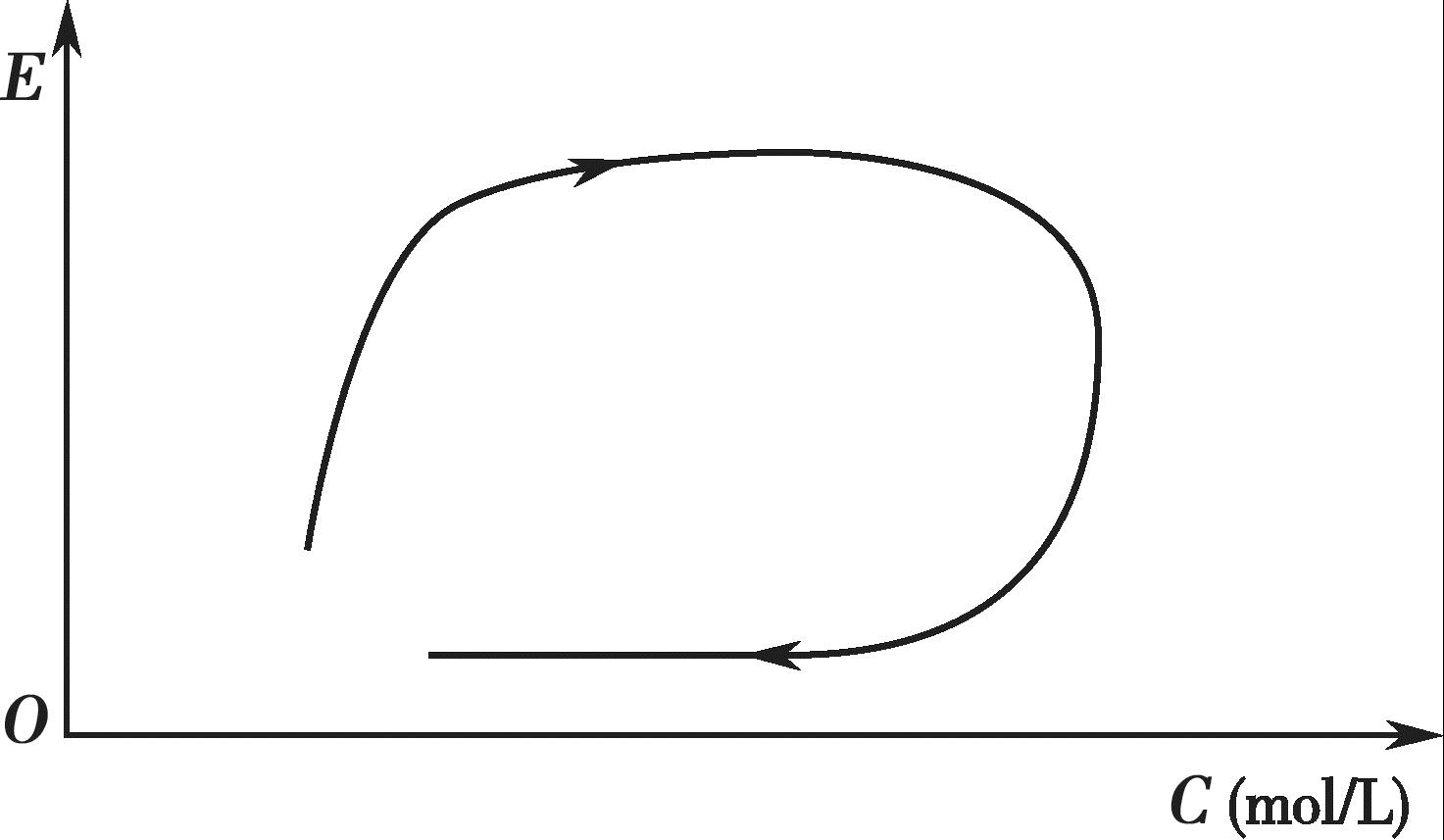
图4-14 血药浓度-效应的顺时针曲线
(3)药理效应部位在不同的房室:
有些药物的药效是由多种药理作用合并产生的,而作用部位可来自同一房室,也可来自不同的房室。如抗高血压药物盐酸可乐定,在低剂量时表现出降压效应,而高剂量时又出现升压效应(图4-15)。为解释这种现象,提出了不同的效应部位,或不同的房室具有不同的受体的概念。
用血压的变化(mmHg)表示药理效应( E ),见图4-15。由图4-15 可知,血药浓度在0.5ng/ml 以下时只有降压效应( E 2 );当血药浓度>0.5ng/ml 时开始出现升压效应( E 1 ),但总体药效仍表现为血压下降;当血药浓度达到大约10ng/ml 时,升压效应和降压效应相等,即 E 1 + E 2 =0,总体药效表现为无效应;而当血药浓度>10ng/ml 时, E 1 > E 2 ,总体药效表现为血压升高。临床上应用可乐定时,应控制给药剂量,使血药浓度不要高于5ng/ml,以免产生升压作用。
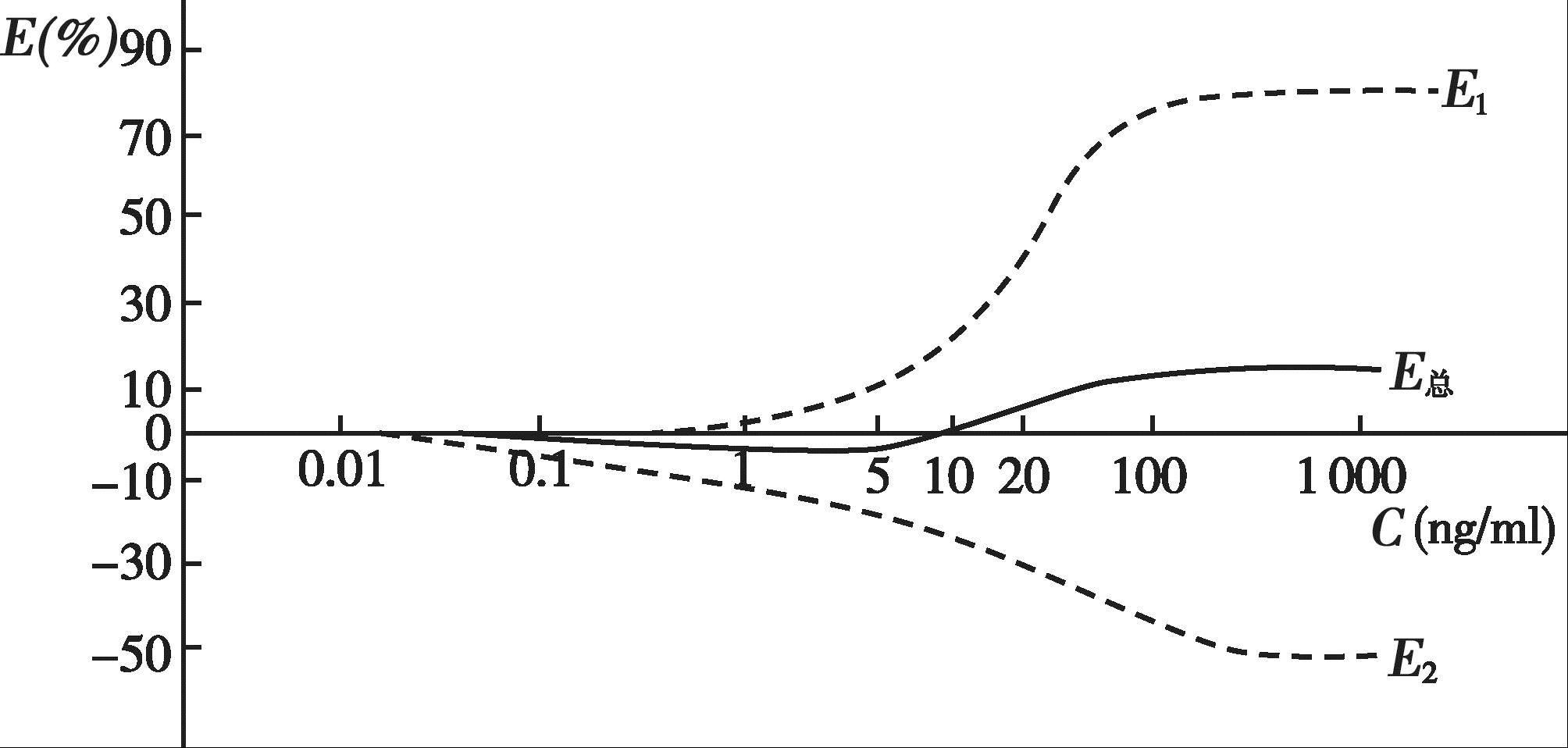
图4-15 可乐定的血药浓度和血压变化(mmHg)曲线
(4)药理效应间接产生:
华法林的药效是间接产生的,如按一定剂量口服给药,尽管华法林的血药浓度由高到低发生波动,但其抗凝作用通常要到2 天后才达到最大活性。这是因为华法林的作用机制是抑制凝血酶原复合物的合成,但不能影响凝血酶原复合物的分解,因此给药后的一段时间内观察不到明显的抗凝活性,直到数天后体内的凝血酶原复合物慢慢分解,才逐渐达到最大的抗凝活性。
若药物的效应与浓度呈直线关系,则可用线性模型来描述两者之间的关系,其表达式为:

式中, E 为效应强度; E 0 为给药前的基础效应; C 为药物浓度; m 是 E 对 C 作直线的斜率。此模型的参数可以通过简单的线性回归求得,它能预报给药前的基础效应是否为0,但不能预报药物的最大效应。线性模型仅适用于浓度较低时, E 的变化正比于 C ,两者存在线性关系。
对数线性模型(log-linear model)是假定药物的效应强度与对数浓度或对数效应强度与对数浓度之间呈直线关系。
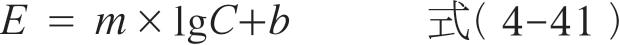
式中, m 、 b 分别为 E 对 C 作半对数直线的斜率、截距。该模型最大的优点在于能够预测相当于最大效应的20%~80%的药物效应强度,但不能预测药物的基础效应和最大效应。
最大效应模型( E max model)描述的是所有血药浓度范围内的浓度-效应关系,即从未用药物时效应为0 到药物浓度远超过EC 50 ( C >>EC 50 )时的最大效应。如果有基线效应( E 0 )存在时,公式可用下式表示:
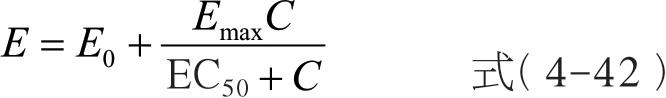
式中, E max 为可能的最大效应;EC 50 为引起50%的最大效应的药物浓度。
药物浓度与效应在最大效应模型中无线性或半对数线性关系。在低浓度时,浓度稍稍变化,效应就有较大的改变;在高浓度时,浓度有较大变化而效应改变不明显。
Sigmoid E max 模型为最大效应模型的扩展,也称为Hill 模型,是目前表述血药浓度与药效强度定量关系时最为常用的模型,公式可用式(4-43)表示。与最大效应模型相比,Hill 模型增加了1 项参数 m ,称为形状因子,其值在1 附近变动,主要反映S 形曲线中间区段斜率的大小。
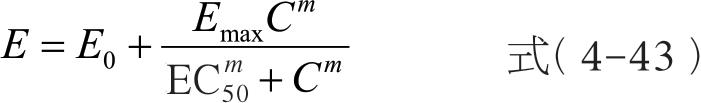
不同的药物具有其特定的EC 50 和 m 值。EC 50 对于估算和确定药物的治疗浓度范围具有指导意义,而 m 值则能反映出药物在一定的浓度范围内其药理效应的变化趋势。当 m =l 时,简化为 E max 模型;当 m <l 时,曲线较为平坦;当 m >1 时,曲线变陡,且更趋向S 形,同时最大效应增大。 m 值越大,曲线越陡,表示效应随浓度变化的幅度越大。如图4-16 所示。
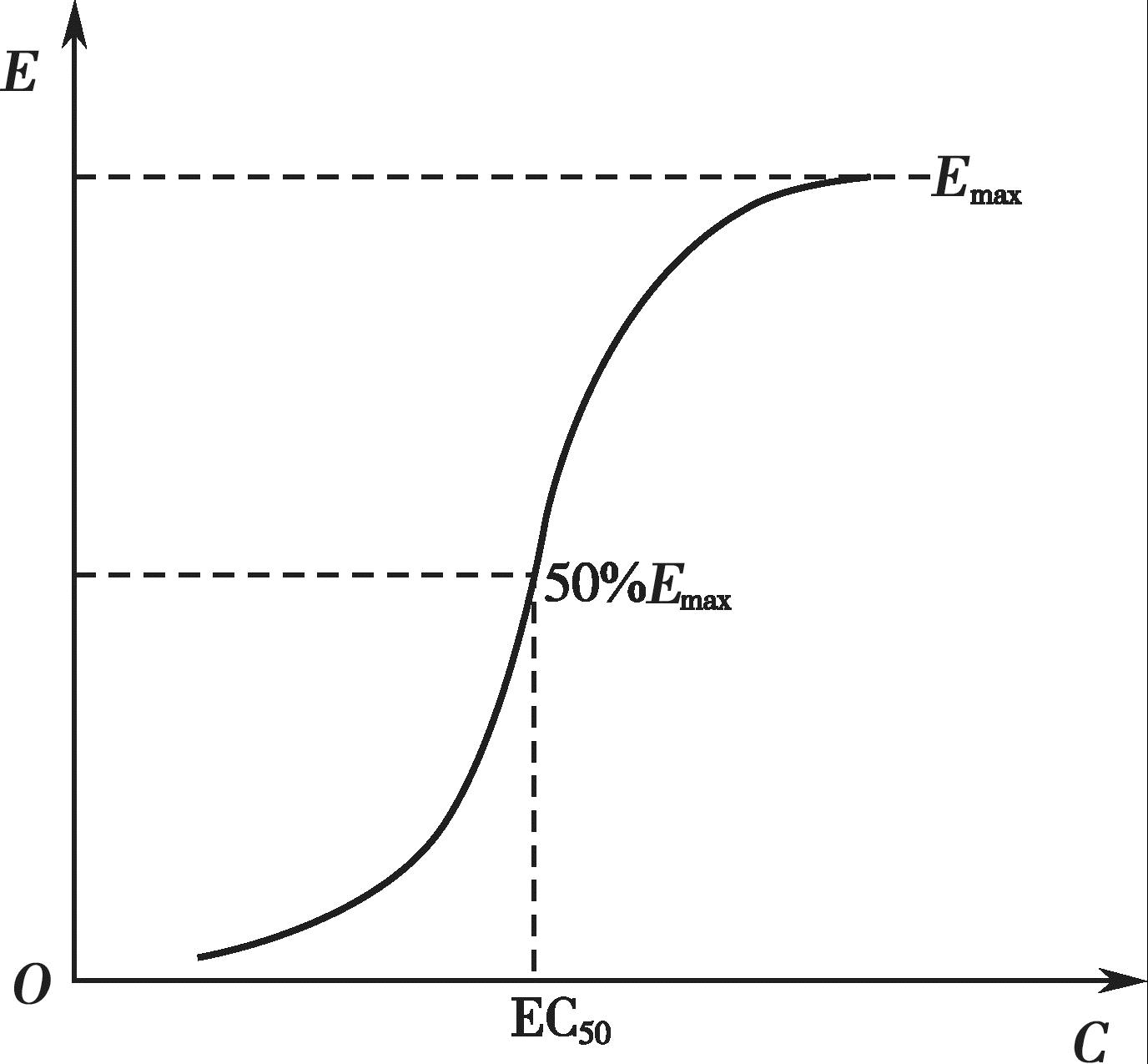
图4-16 S 形 E max 浓度-效应曲线
许多药物药理效应的时间过程与血药浓度-时间过程没有直接的平行关系,最大药理效应可能出现在血药浓度达峰之前或之后。针对这种现象,Sheiner 认为由于药物的作用部位不是血浆,滞后是药物进入和作用于效应部位的结果。1979年,他将经典的药物动力学模型加以扩展,引入了效应室的概念。效应室不是药物动力学模型的一部分,而是与含药物的血浆室连接的虚拟的药效动力学室。药效室的引入,将经典的药动学模型和药效学模型有机地结合了起来,建立了药物动力学-药效动力学结合模型,简称PK-PD 结合模型(图4-17)。Sheiner 等用此模型成功地拟合了筒箭毒碱药效滞后于血药浓度的现象。
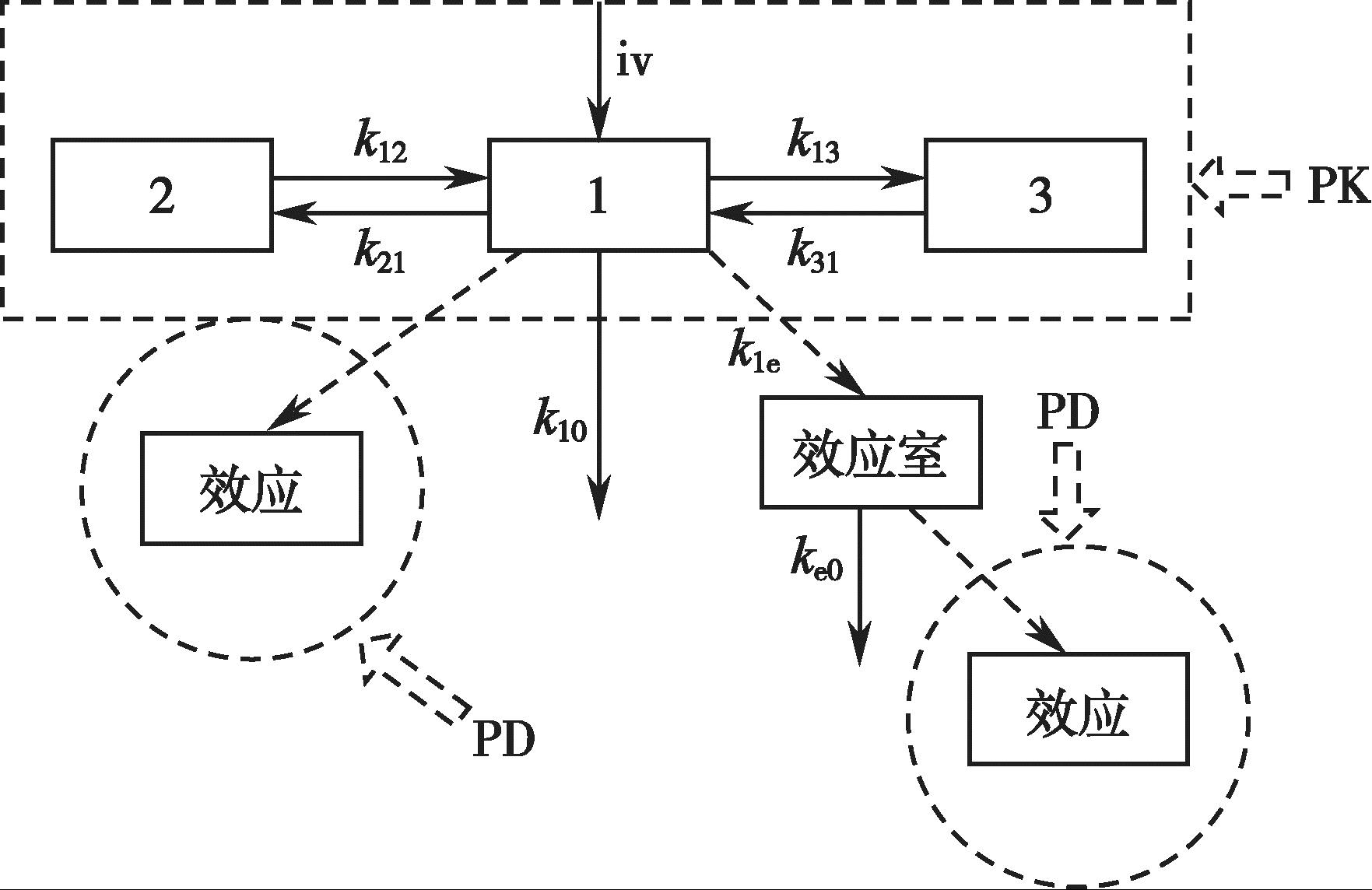
图4-17 PK-PD 模型的示意图
PK-PD 结合模型由图4-17 方框内的药物动力学模型和圆环内的药效动力学模型两部分组成,两部分间通过Hill 方程[式(4-43)]连接。左侧圆环表示药物的作用部位处于中央室或血液灌流充分的组织器官时的药效动力学模型,右侧圆环表示作用部位在药物浓度变化相对滞后于血药浓度的组织或器官时的药效动力学模型。在临床实际用药中,大部分药物的效应达峰时间均明显滞后于血药浓度达峰时间。
药物进入效应室属一级动力学过程,可用一级速率常数 k e0 表示。在血浆浓度恒定的情况下,效应室药物浓度达效应室药物浓度最大值的50%所需要的时间 t 1/2 k e0 =ln2/ k e0 , k e0 越大, t 1/2 k e0 越小,药物峰效应滞后现象越不明显;反之亦然。
有些药物的血药浓度个体差异甚大,加之血药浓度并不与药理效应同步,因此需要建立的PK-PD 结合模型解决药效滞后于血药浓度的现象。建立时间、效应、血药浓度三维统一的关系,这样可达到安全、有效用药的目的。PK-PD 模型突破了简单的药物剂量-效应分析,将药物的特征进一步分为药动学和药效学组分,并对两者进行定量描述。药动学描述机体内剂量和药物浓度随时间变化的关系,药效学则描述药物效应浓度与效应的关系。大部分药物的作用并非在血浆,模型的药动学和药效学必须与效应室联系起来,以便于药物在血浆中的浓度可转化为药物的效应室浓度。PK-PD 模型可用于预测药物峰效应的滞后、幅度和持续时间,为临床麻醉提供合理的剂量、预测作用时间及药物作用强度提供了科学依据,因此是临床麻醉参考的重要依据。
k e0 与效应室浓度及时间的关系见图4-18。
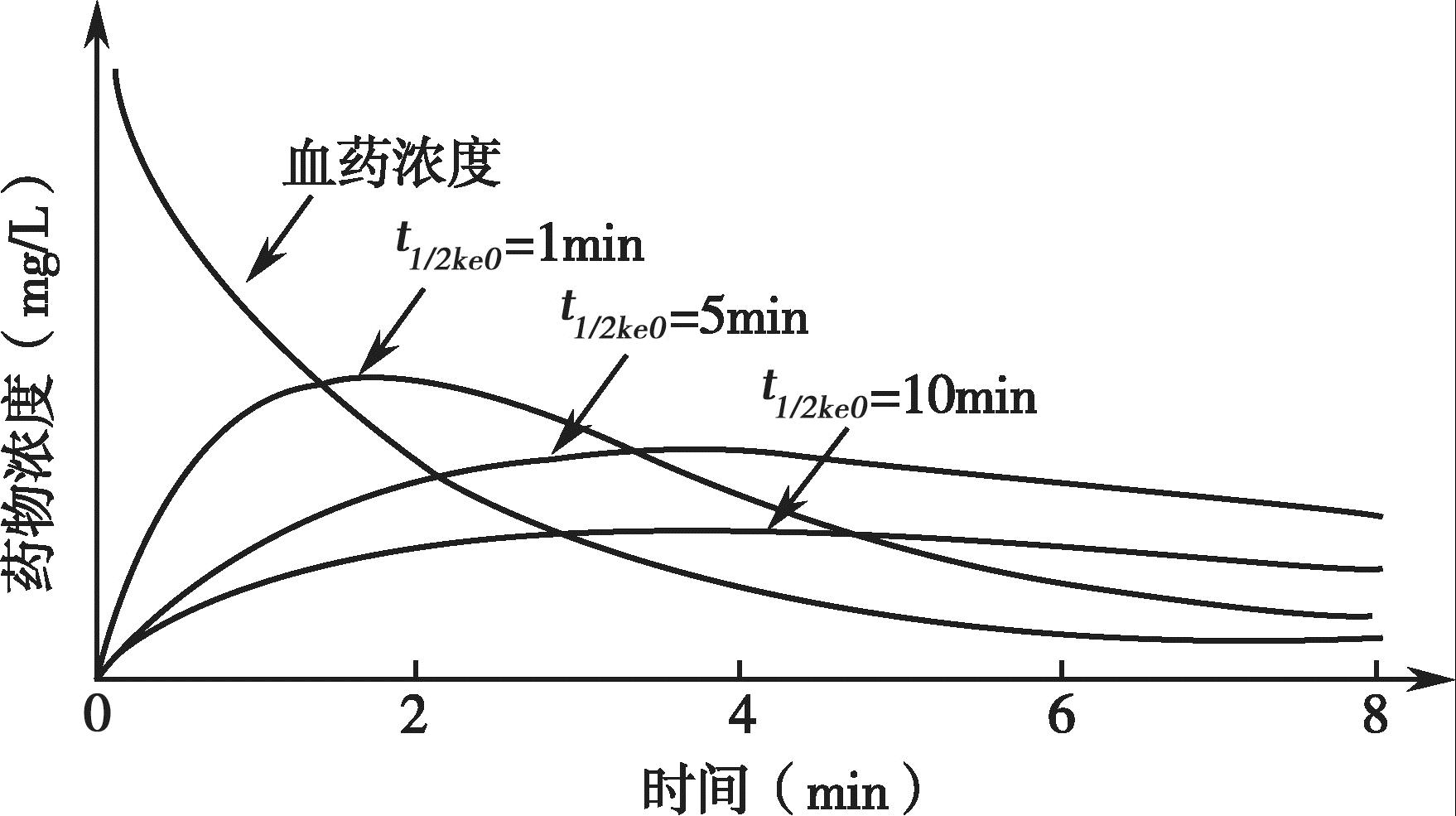
图4-18 k e0 与效应室浓度及时间的关系
比较分析阿片类药物的 k e0 对临床麻醉药的选择很重要。阿芬太尼在中央室与效应室迅速平衡,给予等效剂量药物时,阿芬太尼达峰效应的时间显著快于芬太尼和舒芬太尼,快速起效的阿芬太尼实际上在缓慢起效的芬太尼、舒芬太尼达峰效应前已达到峰效应且效应已经开始下降(图4-19)。
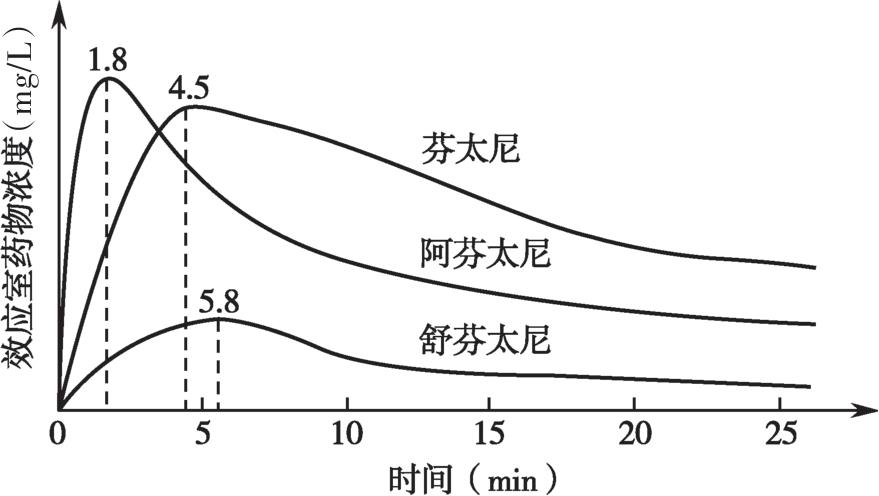
图4-19 等效剂量的芬太尼类药物单次注射后效应室浓度达峰时间的差异
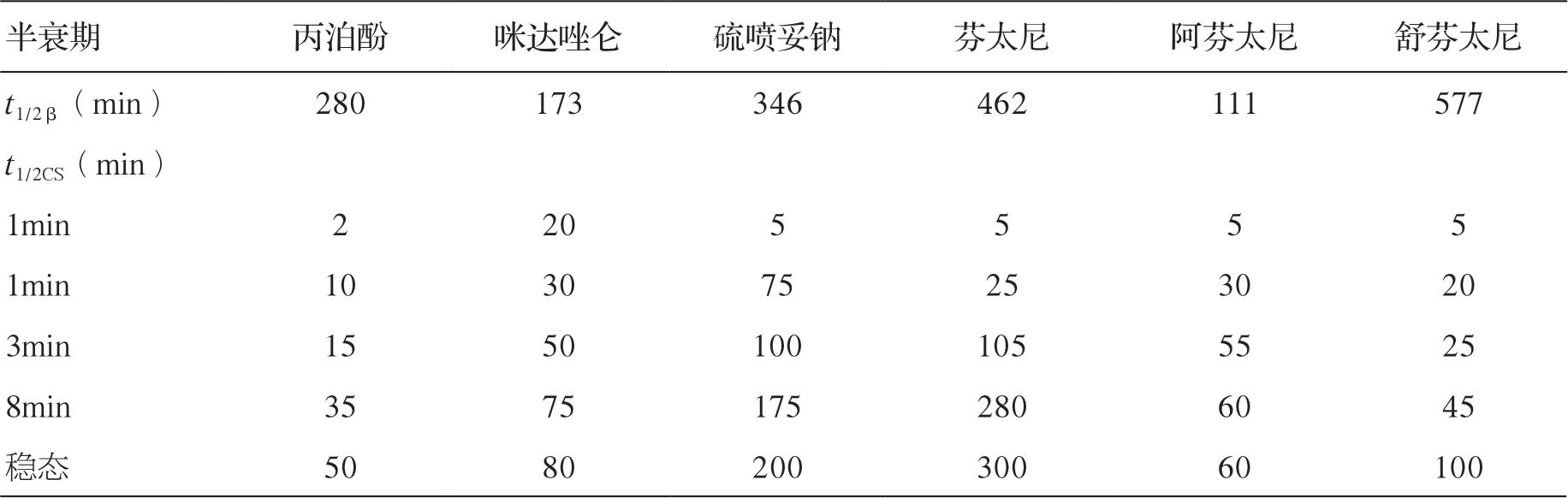
临床麻醉和重症监测中使用的大多数药物的效果和它们的血药浓度直接相关,而停药后药效的消失由血药浓度下降的速度决定。在手术室和ICU,药物常在相对短的时间内使用,远远未能饱和机体的贮存库。目前已清楚短期输注后,药物血浆浓度下降的速度更大程度上是由药物的再分布过程,而不是由实际代谢或消除决定的。例如麻醉医师经常使用硫喷妥钠来进行麻醉诱导,硫喷妥钠的消除半衰期约为12 小时,然而如果不使用其他药物,患者将在几分钟内苏醒。其原因就是再分布过程,即硫喷妥钠由于与脂肪组织的高亲和力,可以从高灌注的组织尤其是中枢神经系统向脂肪转移。时量相关半衰期是指药物停止输注后血液或血浆中的浓度下降50%需要的时间,消除半衰期是指药物代谢所需要的时间,时量相关半衰期不能由消除半衰期来预测,因为它同时依赖于药物的分布。
尽管已经知道药物分布对药效的消失有重要作用,但直至20 世纪80年代末期才认识到此概念的作用。1990年,斯坦福的Shafer和Varvel 两位学者发表了3 种合成阿片类药物(芬太尼、舒芬太尼,阿芬太尼)的模拟药代动力学,研究主要集中在血浆和效应室(CNS)药物浓度的关系上,他们发现在停止药物输注后血浆药物浓度下降20%、50%和80%的时间在很大程度上依赖于药物输注时间的长短,并不能从药物的消除半衰期来预测。因此,学者们使用专业术语时量相关半衰期(contextsensitivity half-time,CSHT)来表示这个参数。在此定义中,context 指药物持续输注的时间长短,此时间与消除半衰期不一致,因此认为时量相关半衰期是停止输注后中央室药动学的有效指标。时量相关半衰期可用单次剂量后的浓度-反应来计算,可通过计算机模拟计算。
另有研究者特别模拟了合成阿片类药物芬太尼、舒芬太尼和阿芬太尼以及催眠镇静药物硫喷妥钠、咪达唑仑和丙泊酚的药动学行为,同样观察到Shafer 和Varvel 的结果,即药物输注的时间长短(context)显著影响药物浓度下降50%所需的时间,而且消除半衰期并不能预测时量相关半衰期。丙泊酚的时量相关半衰期显著短于咪达唑仑,尽管它的消除半衰期(400 分钟)长于咪达唑仑(173 分钟)。
时量相关半衰期和消除半衰期之间的差异反映了药物从高灌注的器官向灌注较差的贮存部位(如肌肉和脂肪,药物在这些部位无相关的生理学效应)再分布过程在药物代谢中的重要性。消除半衰期是从药动学模型得到的一个参数,是药物从机体代谢或清除所需要时间的估计。如果一种药物的药动学可由一室模型很好地描述,那么消除半衰期和时量相关半衰期是相同的。同时,也能预测在药物输注足够长时间满足机体贮存库后,时量相关半衰期和消除半衰期也是相同的。与此一致,研究也显示在长时间的输注后,舒芬太尼(消除半衰期为577分钟)的时量相关半衰期要长于阿芬太尼(消除半衰期为111 分钟)。
常见麻醉药的消除半衰期与时量相关半衰期详见表4-2。
阿芬太尼在持续输注数小时后CSHT 并非最少,舒芬太尼如持续输注时间<8 小时显然具有优越性。根据药动学理论,其间的差异是舒芬太尼有一个大的、缓慢平衡的外周室,停止输注以后药物仍然持续流向外周室,因而中央室浓度下降较快,即输注时间<8 小时时停药,中央室浓度迅速下降是因为消除和分布两者共同作用的结果。芬太尼早期即表现出时间依赖性CSHT 增加,当要求药物浓度快速下降时芬太尼显然不足,而较适用于需维持阿片类药物长期治疗。短时间输注3 种药物的CSHT 近乎相同,因而短期应用时,三药浓度降低50%需要的时间无差异。
图4-20 显示的是血浆浓度下降50%需要的时间与输注时间的关系。
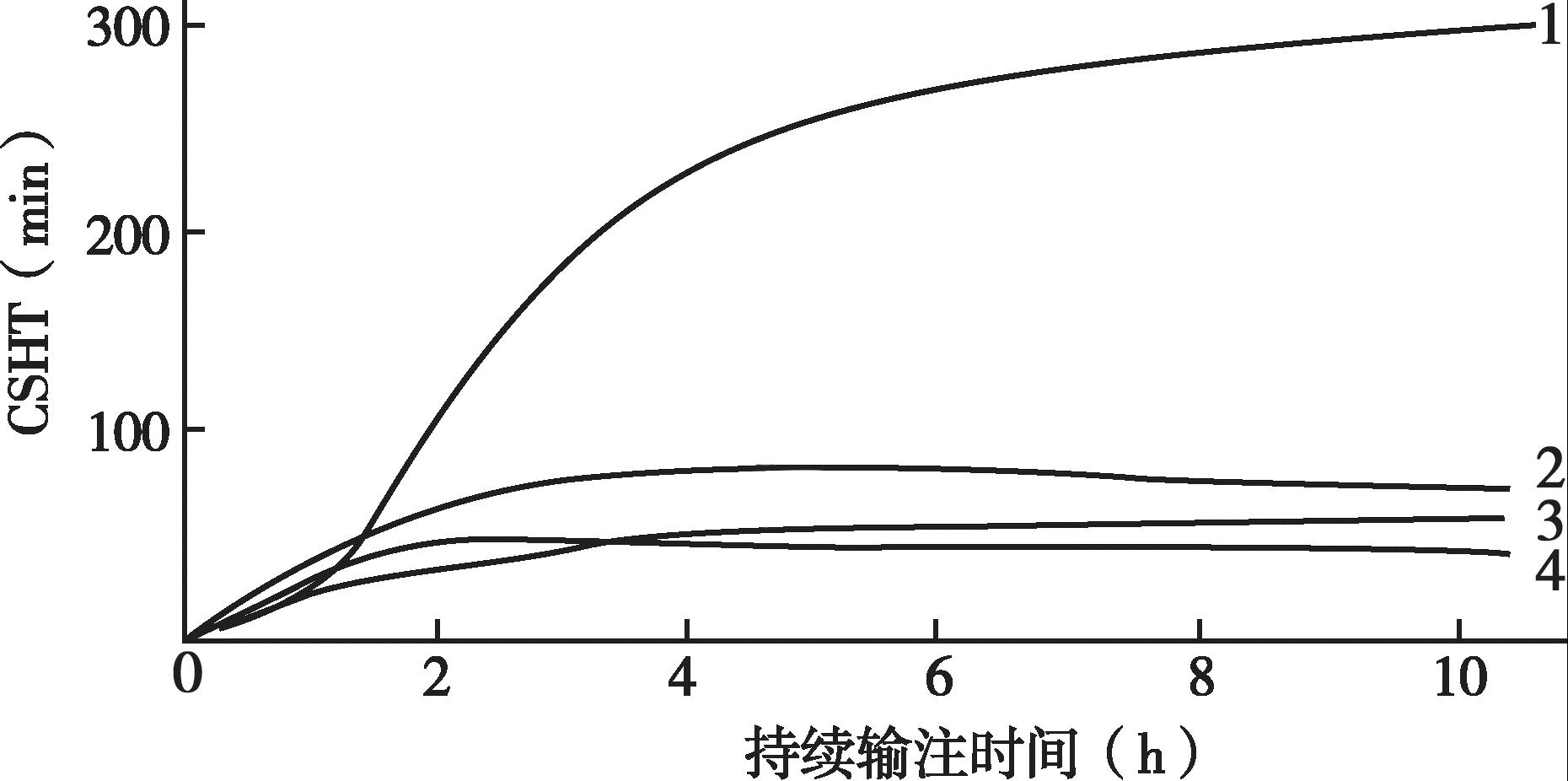
图4-20 血浆浓度下降50%需要的时间与输注时间的关系
1.芬太尼; 2.阿芬太尼;
3.舒芬太尼; 4.丙泊酚。
表4-2 常见麻醉药的消除半衰期与时量相关半衰期
(印晓星)