
下载掌阅APP,畅读海量书库
立即打开



糖尿病的治疗药物主要基于纠正血糖升高的两个主要病理改变:胰岛素抵抗和胰岛素分泌受损。根据给药途径的不同,降血糖药可分为口服类和注射类两种。口服降血糖药可分为以促进胰岛素分泌为主要机制的药物和通过其他机制降低血糖的药物,前者包括磺酰脲类、格列奈类、二肽基肽酶-4(dipeptidyl peptidase-4,DPP-4)抑制剂;后者包括双胍类、噻唑烷二酮类(thiazolidinedione,TZD)、α-葡糖苷酶抑制药、钠-葡萄糖协同转运蛋白2抑制剂(sodium-glucose co-transporter 2 inhibitor,SGLT-2i)。注射类降血糖药包括胰岛素和胰高血糖素样肽-1受体激动剂(glucagon-like peptide 1 receptor agonist,GLP-1RA)。
临床应用时,应依据糖尿病的分型、血糖控制情况、体重、并发症、药品不良反应等因素综合考虑药物的选用。生活方式干预和二甲双胍为2型糖尿病患者高血糖的一线治疗推荐。若无禁忌证,二甲双胍应一直保留在糖尿病的药物治疗方案中。如单药治疗而血糖仍未达标,采用2种甚至3种不同作用机制的药物联合治疗,也可加用胰岛素治疗。合并动脉粥样硬化性心血管疾病(ASCVD)或心血管风险高危的2型糖尿病患者,不论其HbA1c是否达标,只要没有禁忌证,都应在二甲双胍的基础上加用具有ASCVD获益证据的GLP-1RA或SGLT-2i;合并慢性肾脏病(CKD)或心力衰竭的2型糖尿病患者,不论其HbA1c是否达标,只要没有禁忌证,都应在二甲双胍的基础上加用SGLT-2i;合并CKD的2型糖尿病患者,如不能使用SGLT-2i,可考虑选用GLP-1RA。
各类降血糖药的临床应用见下文阐述。
胰岛素可增加葡萄糖的利用,加速葡萄糖的无氧酵解和有氧氧化,促进肝糖原和肌糖原的合成和贮存,抑制糖原的分解和糖异生,因而可使血糖降低。此外,胰岛素还能促进脂肪的合成,抑制脂肪的分解,使酮体的生成减少,纠正酮症酸血症的各种症状;可促进蛋白质的合成,抑制蛋白质的分解。胰岛素和葡萄糖合用,可促使钾从细胞外液进入组织细胞内。胰岛素是治疗糖尿病,特别是1型糖尿病的重要手段。1921年,Fredrick和Charles发现胰岛素的生理作用并将其应用于临床,取得显著疗效。目前,胰岛素制剂的研制开发进展迅速,临床常用的胰岛素品种繁多,可按来源、制备工艺、作用时间等不同进行分类。
根据来源,胰岛素可分为人胰岛素、猪胰岛素和牛胰岛素。
是由胰岛β细胞分泌的激素类物质,是一种小分子蛋白质,分子量为5 808kDa,由A、B两条氨基酸肽链组成。A链有21个氨基酸,B链有30个氨基酸,A、B两链之间以二硫键连接。
是由猪的胰脏提取出来的,与人胰岛素的结构类似,仅有1个氨基酸不同,即人胰岛素B30位的苏氨酸为丙氨酸。
是由牛的胰脏提取出来的,与人的胰岛素有3个氨基酸不同,即人胰岛素B30位的苏氨酸为丙氨酸、A8位的苏氨酸为丙氨酸、A10位的异亮氨酸为缬氨酸。
根据制备工艺,胰岛素可分为动物胰腺提取、半合成及合成人胰岛素和胰岛素类似物。
传统胰岛素是从猪、牛或其混合物中提取的,是一种含有多种生物活性杂质的提取物,现因人胰岛素的广泛使用,已逐渐被淘汰。
是指采用酶修饰、重组DNA技术等制备工艺获得的与人胰岛素的氨基酸序列完全相同的胰岛素。人胰岛素是以猪胰岛素为原料,经过酶修饰后得到的人胰岛素;生物合成人胰岛素是通过重组DNA技术,利用经过基因修饰的细菌产生的人胰岛素。
是指利用重组DNA技术,通过对人胰岛素的氨基酸序列进行修饰生成的、具有胰岛素的功能、可模拟正常胰岛素分泌时相和作用的一类物质。目前,临床常用的有赖脯胰岛素(insulin lispro)、门冬胰岛素(insulin aspart)、谷赖胰岛素(insulin glulisine)、甘精胰岛素(insulin glargine)、地特胰岛素(insulin detemir)、德谷胰岛素(insulin degludec)等。
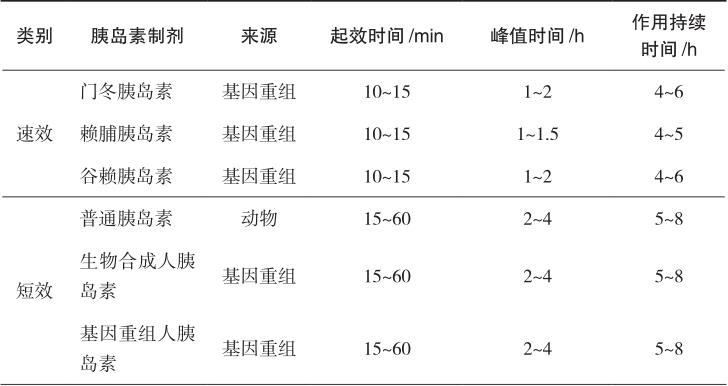
根据作用持续时间,胰岛素可分为速效(超短效)、短效、中效和长效胰岛素,另可将短效和中效胰岛素按不同比例预先混合制成预混胰岛素。临床上常用的胰岛素制剂及作用特点如表2-1所示。
包括门冬胰岛素、赖脯胰岛素、谷赖胰岛素。普通胰岛素是由6个胰岛素分子和2个Zn 2+ 形成的含锌胰岛素六聚体。当普通胰岛素进入血液中时,立即解离成单体,并能够与胰岛素受体结合,静脉注射后立即发挥降糖作用。但当普通胰岛素注射到皮下组织中时,六聚体必须分解为二聚体或单体吸收入血才能起效。因此,普通胰岛素皮下注射时降糖作用会延迟,一般需餐前30分钟注射。速效胰岛素类似物是适当改变人胰岛素分子的一级结构,使之达到速效的目的。门冬胰岛素是将人胰岛素B28位的脯氨酸由天冬氨酸代替,赖脯胰岛素是将人胰岛素B28位的脯氨酸和B29位的赖氨酸的位置互换,谷赖胰岛素是以赖氨酸替代人胰岛素B3位的天冬氨酸并以谷氨酸替代人胰岛素B29位的赖氨酸。这些经过修饰的胰岛素类似物降低自我聚合能力,不再容易形成六聚体结晶,而是以单体的形式存在,皮下注射后吸收快,10~15分钟起效,1~2小时作用达峰值,作用持续4~6小时。该类胰岛素作为餐时胰岛素使用,可更好地模拟人体生理性分泌的胰岛素作用模式,控制餐后血糖并减少低血糖的发生。该类胰岛素的用药时间灵活,餐前或餐后立刻给药可达到与餐前30分钟注射普通胰岛素的相同的降糖效果,提高患者的依从性。
包括普通胰岛素、生物合成人胰岛素和基因重组人胰岛素。常用的生物合成人胰岛素制剂有诺和灵R(Novolin R);基因重组人胰岛素制剂有优泌林R(Humulin R)、甘舒霖R及重和林R等。此类胰岛素为无色透明液体,可供皮下注射或静脉注射。皮下注射后0.5~1小时开始起效,2~4小时达到作用高峰,作用持续5~8小时。
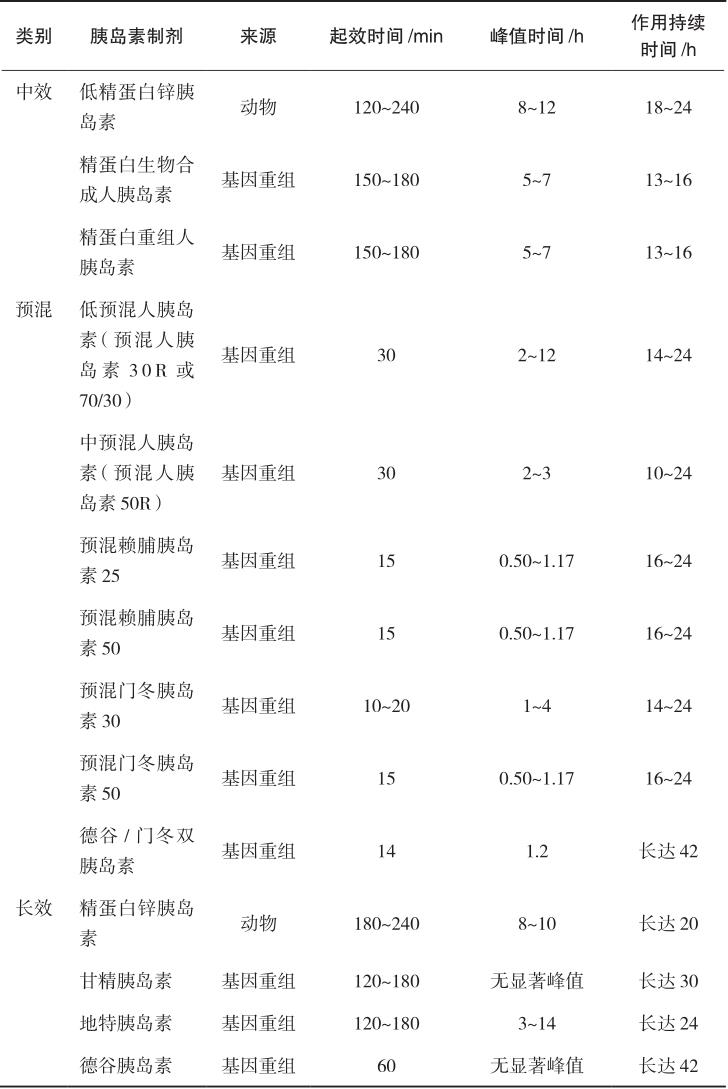
中效胰岛素又称低精蛋白锌胰岛素(NPH),是在胰岛素中加入等量的鱼精蛋白和微量锌,注射后在局部逐渐溶解,缓慢释放出胰岛素而被人体吸收。常用的制剂有精蛋白生物合成人胰岛素、精蛋白重组人胰岛素等。该类制剂为混悬液,只能皮下注射或肌内注射,不可静脉注射,主要用于空腹血糖控制不佳者,可与短效胰岛素或口服降血糖药联合使用。皮下注射后2~4小时起效,6~12小时达到作用高峰,作用持续16~24小时。
是指为满足临床对餐后血糖、空腹血糖控制良好和减少注射次数的需要,将速效/短效与中效胰岛素预先混合制备的胰岛素制剂,主要包括预混人胰岛素和预混胰岛素类似物。预混人胰岛素是将重组人胰岛素和精蛋白锌重组人胰岛素按一定比例混合制备的胰岛素制剂,包括低预混人胰岛素和中预混人胰岛素。低预混人胰岛素为30%的短效+70%的中效,如诺和灵30R、优泌林30R、重和林M30、甘舒霖30R等;中预混人胰岛素为50%的短效+50%的中效,如诺和灵50R、甘舒霖50R等。该类胰岛素皮下注射后约0.5小时开始起效,预混人胰岛素2~3小时达到作用高峰,作用可持续14~24小时。预混胰岛素类似物是将速效胰岛素类似物与精蛋白锌胰岛素类似物按一定比例混合而成的胰岛素制剂,包括低预混胰岛素类似物和中预混胰岛素类似物。在国内上市的低预混胰岛素类似物包括优泌乐25(25%赖脯胰岛素+75%精蛋白锌赖脯胰岛素)、诺和锐30(30%门冬胰岛素+70%精蛋白锌门冬胰岛素);中预混胰岛素类似物包括优泌乐50(50%赖脯胰岛素+50%精蛋白锌赖脯胰岛素)、诺和锐50(50%门冬胰岛素+50%精蛋白锌门冬胰岛素)等。与预混人胰岛素相比,预混胰岛素类似物在血糖控制尤其是餐后血糖控制和减少低血糖发生方面更具优势。该类胰岛素可于餐前或进餐后立刻皮下注射,预混胰岛素类似物注射后15分钟开始起效,30~70分钟达到作用高峰,作用可持续16~24小时。
长效胰岛素又称精蛋白锌胰岛素,是在胰岛素中加入过量的鱼精蛋白与锌离子,作用时间进一步延长,皮下注射后3~4小时起效,8~10小时达峰,药效持续长达20小时。长效胰岛素类似物包括甘精胰岛素、地特胰岛素、德谷胰岛素,是在人胰岛素的基础上经过分子修饰,皮下注射后形成多聚体,从而延长药物的吸收和作用时间。相对于长效胰岛素,长效胰岛素类似物能更好地模拟生理性胰岛素分泌,作用时间长,无显著的峰值,作用平稳,降低低血糖的发生风险。
表2-1 常用的胰岛素制剂及作用特点
续表
非胰岛素类降血糖药主要有磺酰脲类药物、格列奈类药物、双胍类药物、噻唑烷二酮类药物、α-葡糖苷酶抑制药和二肽基肽酶-4抑制剂、钠-葡萄糖协同转运蛋白2抑制剂,注射制剂有胰高血糖素样肽-1受体激动剂。具体分类与特点如下。
本类药物有2种,即苯乙双胍和二甲双胍。苯乙双胍导致乳酸酸中毒的风险较大,已不在临床使用。目前,临床上使用的双胍类药物主要是二甲双胍。因二甲双胍不仅有确切的降血糖疗效,还可降低体重、保护心血管系统,在国内外的糖尿病诊治指南中,二甲双胍被推荐为2型糖尿病患者控制高血糖的一线药物和联合用药的基本药物。二甲双胍不仅是超重或肥胖的2型糖尿病患者的首选药,也适用于体重正常的2型糖尿病患者,同时可与胰岛素联合治疗1型糖尿病。双胍类药物降糖作用的主要机制包括:①作用于肝脏,抑制糖异生,减少肝糖输出;②作用于脂肪、肌肉组织,提高机体对胰岛素的敏感性,增加机体对葡萄糖的摄取和利用,促进糖原合成,降低游离脂肪酸;③作用于肠道,抑制葡萄糖的吸收,提高GLP-1水平。二甲双胍的减重作用机制可能包括抑制食欲,减轻热量摄入;改善高胰岛素血症;增加机体对瘦素的敏感性。另外,二甲双胍能改善脂肪的合成与代谢,可降低2型糖尿病患者的血浆甘油三酯、低密度脂蛋白胆固醇水平,对高密度脂蛋白水平的影响不明显;对非酒精性脂肪肝患者的肝脏血清酶谱及代谢异常均有显著改善。
二甲双胍的降糖作用具有剂量依赖效应,起效最小剂量为500mg/d,最佳有效剂量为2 000mg/d,成人的最大推荐剂量为2 550mg/d。使用时的剂量调整原则为“小剂量起始,逐渐加量”,即开始服用500mg/d或<1 000mg/d,1~2周后加量至最大有效剂量2 000mg/d或最大耐受剂量。口服生物利用度为50%~60%,达峰时间约为2.5小时,在血浆中几乎不与血浆蛋白结合,不经肝脏代谢,以原型随尿液排出,血浆半衰期约为6.5小时。虽然血浆半衰期较短,但因其同时分布于红细胞内,使得全血消除半衰期达17.6小时,因此,二甲双胍的普通制剂每次1 000mg,每日2次,可维持24小时的有效血药浓度。由于本品主要以原型由肾脏排泄,清除迅速,12~24小时可清除90%,肾功能减退患者需根据估算的肾小球滤过率(eGFR)调整给药剂量。
肝功能正常者服用推荐剂量范围的二甲双胍不会造成肝损害,但严重肝损害会明显限制乳酸的清除能力,血清氨基转移酶轻度升高的患者使用时应密切监测肝功能,血清氨基转移酶超过3倍健康人群高限或有严重肝功能不全患者应避免使用二甲双胍。经肾小球滤过和肾小管分泌是二甲双胍清除的主要途径,肾脏清除率约为肌酐清除率的3.5倍,目前尚无确切的证据证明二甲双胍的使用与乳酸酸中毒有关,肝、肾功能正常者长期应用并不增加乳酸酸中毒的风险。肾损害时易发生二甲双胍与乳酸的体内蓄积,有可能会增加乳酸酸中毒的风险,eGFR<45ml/(min·1.73m 2 )和低氧血症患者不建议使用。
磺酰脲类药物属于胰岛素促泌剂,目前在我国上市的有格列本脲、格列齐特、格列吡嗪、格列喹酮、格列美脲等。
磺酰脲类药物通过特异性地结合胰岛β细胞膜上的磺酰脲受体(SUR),使K + 通道关闭,细胞内的K + 外流减少,导致细胞膜去极化,使电压依赖性Ca 2+ 通道开放,细胞外的Ca 2+ 内流而触发胰岛素的释放。人体不同组织的SUR存在异构体,SUR有SUR1和SUR2两个类型,SUR2又可分为SUR2A和SUR2B。SUR1存在于胰岛β细胞膜;SUR2A存在于心肌细胞,SUR2B存在于平滑肌细胞。不同磺酰脲类胰岛素促泌剂与不同SUR类型的结合力存在差异。有研究表明,格列齐特、格列美脲、格列吡嗪对SUR1的选择性较强,而对SUR2A和SUR2B的作用较弱;而格列本脲对SUR1、SUR2A和SUR2B都有作用,临床使用时需注意可能会影响心肌缺血预适应。格列本脲和格列美脲与SUR1的不同亚单位特异性结合,格列本脲与140kDa受体蛋白结合,而格列美脲与65kDa受体蛋白结合,且结合快,解离也快,可快速起效,较少发生低血糖。与其他磺酰脲类药物相比,格列美脲对心血管系统的影响更小,具有一定的胰岛素增敏作用。
磺酰脲类促泌剂品种繁多,20世纪50年代开发的第一代磺酰脲类降血糖药以甲苯磺丁脲、氯磺丙脲为代表,因其服药量大、降血糖持续时间长、存在严重而持久的低血糖反应等,现已退出市场。第二代磺酰脲类降血糖药包括格列本脲、格列吡嗪、格列齐特和格列喹酮等,与第一代磺酰脲类降血糖药相比,第二代药物对受体的亲和力高,降糖活性高,口服吸收快,且引发低血糖、粒细胞减少及心血管不良反应的概率较小。除降糖作用外,格列齐特和格列吡嗪还有减少血小板聚集、改善血液黏度和微循环的作用,有利于减轻或延缓糖尿病血管并发症的发生。第三代磺酰脲类药物格列美脲具有与SUR特异性结合、结合快、解离快、可快速起效、较少发生低血糖等优点,且具有增加胰岛素敏感性、改善胰岛素抵抗的作用。消渴丸是含有格列本脲和多种中药成分的固定剂量复方制剂,降糖效果与格列本脲相当。
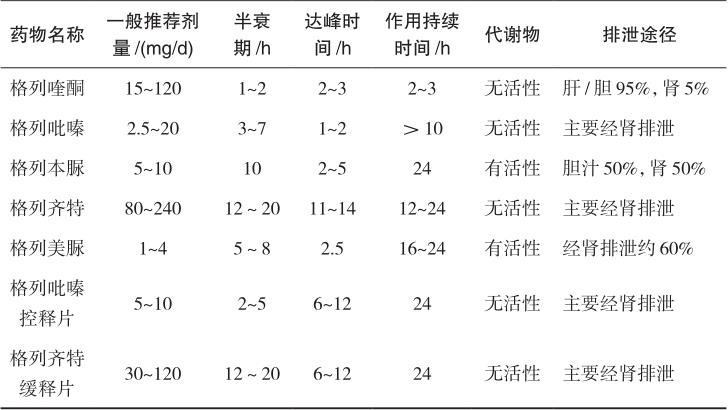
不同磺酰脲类药物的药效学和药动学存在较大的差异,表2-2中列出常用磺酰脲类药物品种和剂型的作用特点。根据作用时间,分为短效制剂和中、长效制剂。短效制剂的半衰期短,作用迅速,主要用于控制2型糖尿病的餐后血糖,包括格列喹酮、格列吡嗪等。格列喹酮的大部分代谢产物由胆汁入肠道,仅5%经肾脏排泄,可用于轻至中度肾功能不全者。中、长效制剂的半衰期长,作用较持久,用于控制2型糖尿病患者的空腹血糖和餐后血糖,包括自身半衰期较长的普通剂型如格列本脲和格列美脲,以及剂型改良之后的缓控释制剂,如格列齐特缓释片和格列吡嗪控释片。格列齐特缓释片采用以亲水性羟丙甲纤维素为基质的缓释技术,格列吡嗪控释片通过胃肠道治疗系统(GITS)技术实现控释,保证全天血药浓度平稳,更符合2型糖尿病24小时基础血糖的控制要求,较少引起低血糖。
磺酰脲类药物为2型糖尿病患者的常用药品,但其降糖作用依赖于一定的胰岛β细胞功能,因此磺酰脲类药物对临床上新诊断的2型糖尿病患者一般十分有效。对于临床诊断超过10年的2型糖尿病患者,由于其胰岛β细胞功能随时间而衰减,磺酰脲类药物的疗效相对较差。磺酰脲类药物常见的不良反应是低血糖和体重增加,特别是老年患者或者肝、肾功能不全的患者,用药期间发生低血糖的风险更高。
表2-2 常用磺酰脲类药物的作用特点
格列奈类药物和磺酰脲类药物的作用机制有相同之处,均与胰岛β细胞膜SUR结合,使K + 通道关闭,导致细胞膜去极化,细胞外的Ca 2+ 内流而促发胰岛素的释放。与磺酰脲类药物不同的是,此类药物与胰岛β细胞K ATP 通道上的36kDa受体蛋白结合而发挥作用,具有“快开-快闭”的特性。其“快开”作用可有效地促进早时相胰岛素分泌,与食物引起的生理性早时相胰岛素分泌相似;而其“快闭”作用不会同时导致基础或第二相胰岛素的过度分泌。这种“快开-快闭”的特性可有效地控制餐后血糖升高,又防止对胰岛β细胞的过度刺激,能够预防高胰岛素血症,减少低血糖的发生。另外,格列奈类药物的促胰岛素分泌作用部分通过葡萄糖介导,依赖于一定浓度的血浆葡萄糖水平。与空腹状态相比,进食前10分钟给药时,格列奈类药物的促胰岛素分泌作用显著增强。
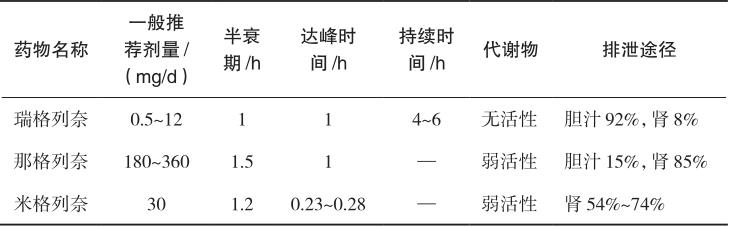
格列奈类促泌剂包括瑞格列奈、那格列奈和米格列奈,其作用特点如表2-3所示。此类药物口服吸收迅速,达峰时间<1小时,不同格列奈类药物的吸收速率和生物利用度受食物的影响程度不同。瑞格列奈的吸收受食物影响较小;那格列奈在肠道内通过那格列奈/H + 共同转运系统快速吸收,其吸收在肠道转运受饮食影响,餐前服药比餐后服药产生的血药浓度峰值高,吸收迅速而完全;米格列奈餐前5分钟内服用,给药后0.23~0.28小时达到最高血药浓度,半衰期约为1.2小时。格列奈类药物大部分与血浆蛋白结合,蛋白结合率为97%~99%。瑞格列奈和那格列奈的代谢主要通过有机阴离子转运多肽OATP1B1转运和肝微粒体细胞色素P450同工酶系统完成生物转化,肝细胞色素P450是体内参与药物代谢的重要酶系,其活性受到诱导或抑制后将干扰药物的作用。因此,瑞格列奈和那格列奈与多种药物存在相互作用。米格列奈经UGT1A9和UGT1A3代谢,极少量经CYP2C9代谢。瑞格列奈及其代谢物92%通过胆汁排泄,8%经尿排泄,可用于肾功能不全患者。
表2-3 格列奈类促泌剂的作用特点
α-葡糖苷酶抑制药主要作用于小肠上皮刷状缘,竞争性地抑制α-葡萄糖苷酶的活性,防止1,4-糖苷键水解,使多糖、双糖的消化延缓,水解产生葡萄糖的速度减慢,延缓单糖的吸收,降低餐后血糖峰值。α-葡糖苷酶抑制药可用于缓解糖尿病患者的餐后高血糖,使血糖高峰与低谷之间的间距缩短,适用于糖耐量减低阶段、糖尿病早期以及以碳水化合物为主要食物成分和以餐后血糖升高为主的患者。本药对葡糖苷酶有高度亲和性,延缓肠内双糖、低聚糖和多糖的吸收,使餐后血糖水平上升被延迟或减弱,拉平昼夜的血糖曲线,减少血糖的大幅波动。
目前,临床应用的α-葡糖苷酶抑制药有阿卡波糖、伏格列波糖和米格列醇。给药应从小剂量开始,逐渐增加剂量,于进餐时随第一口主食一起嚼碎后服用。阿卡波糖的起始剂量为50mg/次,3次/d,根据餐后血糖逐渐增加用药剂量,一般最大剂量为300mg/d。伏格列波糖的起始剂量为0.2mg/次,3次/d,根据餐后血糖可增至0.3mg/次。米格列醇的起始剂量为25mg/次,3次/d;维持剂量为50mg/次,3次/d;最大剂量不超过100mg/次。口服阿卡波糖有1%~2%的活性抑制剂经肠道吸收,未发现在体内有可测定的代谢现象;相反,在肠腔内阿卡波糖被消化酶和肠道细菌分解,其降解产物可于小肠下段被吸收。由于阿卡波糖只作用于肠道,所以在体内的生物利用度极低,但低的生物利用度与治疗效果无关。
α-葡糖苷酶抑制药可单独使用,也可与胰岛素、胰岛素促泌剂、双胍类合并使用。单独服用本类药物通常不会发生低血糖,并可降低餐前反应性低血糖的发生风险,但如与促泌剂或胰岛素合用,有发生低血糖事件的报道。服用α-葡糖苷酶抑制药期间出现的低血糖应使用葡萄糖或蜂蜜予以纠正,食用蔗糖或淀粉等食物矫正低血糖的效果较差。
TZD的作用机制与特异性结合并激活过氧化物酶体增殖物激活受体-γ(PPARγ)有关。PPARγ被激活后,调控与胰岛素效应有关的多种基因的转录,这些基因的功能涉及葡萄糖的产生、转运、利用以及脂肪代谢的调节。其作用表现为:①增强机体对胰岛素的敏感性,加强胰岛素所引起的葡萄糖转运蛋白(GLUT)向细胞内转运,促使胰岛素介导的葡萄糖摄取,增加肌肉、肝脏和脂肪组织对胰岛素的敏感性,减轻胰岛素抵抗;②增加肝糖原合成酶的活性,减少肝内糖异生;③降低血浆甘油三酯、脂肪酸水平,升高高密度脂蛋白水平;④减少尿蛋白的排泄。
目前,在我国上市的TZD有罗格列酮和吡格列酮。这类药物口服吸收迅速,几乎完全吸收,进食对吸收总量无明显影响,但达峰时间延迟,血浆蛋白结合率>99%。罗格列酮部分经CYP2C8代谢为有微弱活性的代谢产物,64%以原型经肾脏排出体外, t 1/2 为3~4小时;吡格列酮主要经CYP2C8和CYP3A4代谢为活性代谢产物,由胆汁排泄, t 1/2 为3~7小时,总吡格列酮(吡格列酮和其活性代谢产物)的 t 1/2 为16~24小时。此类药物起效缓慢,如罗格列酮需要治疗8~12周后再评价疗效和进行剂量调整。
TZD可单独使用,也可与胰岛素、胰岛素促泌剂、双胍类联合使用,用于2型糖尿病,尤其适用于高胰岛素血症或胰岛素抵抗患者,可明显降低空腹血糖及胰岛素水平,对餐后血糖和胰岛素亦有降低作用。单独使用时不导致低血糖,但与胰岛素或胰岛素促泌剂联合使用时可增加低血糖的发生风险。TZD可能增加骨折和心力衰竭的发生风险,有心力衰竭(心功能不全Ⅱ级以上)、活动性肝病或氨基转移酶升高超过健康人群高限2.5倍以上以及严重骨质疏松的患者应禁用此类药物。
GLP-1RA为肠促胰素类药物,可通过模拟天然GLP-1,激活其受体发挥作用,可使GLP-1浓度达到药理水平。肠促胰素是在进食刺激下由肠道细胞分泌的激素,可调节胰岛素对进食的反应,其引起的胰岛素分泌能力占全部胰岛素分泌量的50%~70%,在血糖调节中发挥重要作用。糖依赖性胰岛素释放肽(GIP)和GLP-1是2种主要的肠促胰素。GIP在碳水化合物和脂质的刺激下,主要由十二指肠和空肠近端的K细胞分泌,与胰岛β细胞上的特异性受体结合,促进胰岛素分泌。但2型糖尿病患者的循环GIP正常或升高,同时GIP对胰岛β细胞的促胰岛素分泌作用显著降低,对胰岛α细胞也没有作用,因而限制了其临床应用。GLP-1在食物刺激下由回肠和结肠的L细胞分泌入血,其不仅可增强胰岛β细胞反应,还能通过作用于胰岛α细胞减少胰高血糖素分泌,进而减少肝糖输出,以及作用于进食中枢和胃,抑制食欲并减缓胃排空,从而降低胰岛β细胞负荷,对2型糖尿病患者的代谢异常进行多个方面的调控。由于生理性分泌的GLP-1在体内迅速被二肽基肽酶-4(DPP-4)降解,限制了其作用时间。GLP-1RA通过激动GLP- 1受体,起到和内源性GLP-1类似的作用。而且与天然GLP-1的不同之处是,GLP-1RA不容易被DPP-4降解,延长了半衰期,增加了活性GLP-1在体内的浓度。
目前,国内已上市的GLP-1RA有艾塞那肽、利拉鲁肽、贝那鲁肽、利司那肽、度拉糖肽、艾塞那肽微球和聚乙二醇洛塞那肽,均需皮下注射。其中短效制剂包括艾塞那肽、贝那鲁肽及利司那肽,一般需要每日1~3次皮下注射;长效制剂包括利拉鲁肽,需要每日1次皮下注射;超长效制剂包括度拉糖肽、艾塞那肽周制剂及聚乙二醇洛塞那肽,一般需要每周1次皮下注射。GLP-1RA用于2型糖尿病患者,可单独使用或与其他口服降血糖药联合使用。不仅可有效降低血糖,兼具低血糖发生率低的优点,还具有降低体重、收缩压及改善血脂紊乱的作用。
生理性分泌的GLP-1在体内迅速被DPP-4降解,DPP-4i通过抑制DPP-4而减少GLP-1在体内的失活,使内源性GLP-1水平升高。GLP-1以葡萄糖浓度依赖性方式增强胰岛素分泌,抑制胰高血糖素分泌,发挥降低糖化血红蛋白、空腹血糖和餐后血糖的作用。需要注意的是,DPP-4i降低HbA1c的程度与基线HbA1c水平有一定的关系,即基线HbA1c水平高的降低幅度明显。
目前,国内已上市的DPP- 4抑制剂有西格列汀、沙格列汀、维格列汀、利格列汀和阿格列汀。DPP-4抑制剂适用于成人2型糖尿病患者的血糖控制,可单独使用或与二甲双胍联合使用。DPP-4抑制剂均为口服制剂,使用方便,且具有呈葡萄糖浓度依赖性地降低血糖的作用,低血糖发生率低,对体重的影响小。单独使用DPP-4抑制剂不增加低血糖发生的风险,DPP-4抑制剂对体重的作用为中度或轻度增加。在有肾功能不全的患者中使用西格列汀、沙格列汀、阿格列汀和维格列汀时,应注意按照药品说明书来减少药物剂量。肝、肾功能不全的患者使用利格列汀时不需要调整剂量。
目前在我国被批准临床使用的SGLT-2i为达格列净、恩格列净和卡格列净。SGLT-2i通过抑制肾脏肾小管中负责从尿液中重吸收葡萄糖的SGLT-2降低肾糖阈,促进尿葡萄糖排泄,从而达到降低血液循环中葡萄糖水平的作用。SGLT-2i与其他口服降血糖药比较,其降糖疗效与二甲双胍相当。在具有心血管高危风险的2型糖尿病患者中应用恩格列净或卡格列净的临床研究结果显示,该类药物可使主要心血管不良事件和肾脏事件复合终点发生发展的风险显著下降,心衰住院率显著下降。SGLT-2i单独使用时不增加低血糖发生的风险,联合胰岛素或磺酰脲类药物时,可增加低血糖发生风险。SGLT-2i在中度肾功能不全的患者中可以减量使用。在重度肾功能不全患者中因降糖效果显著下降不建议使用。SGLT-2i的不良反应包括直立性低血压、生殖泌尿道感染、酮症酸中毒等。