
下载掌阅APP,畅读海量书库
立即打开



补体系统是广泛存在于人和动物体液及细胞表面的一组蛋白,可由肝细胞、巨噬细胞及肠黏膜上皮细胞等多种细胞产生。它由近40种成分组成,包括13种固有成分(C1q、C1r、C1s、C2-C9、D因子及B因子),多种调节蛋白(备解素、H因子、I因子、衰变加速因子等)以及补体受体等。其中C3为含量最高的成分。补体需活化后才能发挥生物学效应,其活化过程表现为一系列丝氨酸蛋白酶的级联酶解反应。
补体活化主要通过3条途径(图6-2-1),即经典途径,旁路途径和凝集素(MBL)途径。其中经典途径是含IgG(IgG1-3)或者IgM的免疫复合物被C1q识别并与之结合。随后C1qrs组装并切割C2、C4形成C3转化酶(C4b2a)。C3转化酶可将C3切割成C3a和C3b,后者将C3转化酶进一步形成C5转化酶。同样的,旁路途径通过C3自发的水解,形成C3Bb,后者将C3裂解成C3a及C3b。该途径的扩增基于C3b与活化表面(例如细菌表面)的共价结合,然后再切割因子B,在因子D和备解素的存在下,形成C3转化酶(C3bBb)。凝集素途径的激活则基于通过甘露聚糖凝集素或纤维胶凝蛋白识别微生物细胞表面碳水化合物。该过程导致C2和C4的切割以形成经典途径C3转化酶(C4bC2a)。膜攻击为所有途径发挥作用的共同通路。C5被C5转化酶裂解成C5a及C5b,后者与C6、C7、C8、C9结合形成C5b-9,即膜攻击复合物(membrane attack complex,MAC)。MAC可插入细胞膜的脂质双层引起细胞裂解。对于肾实质细胞,C5b-9的量很少,通常不足以诱导细胞裂解,但这些亚溶解量仍然可导致细胞损伤。
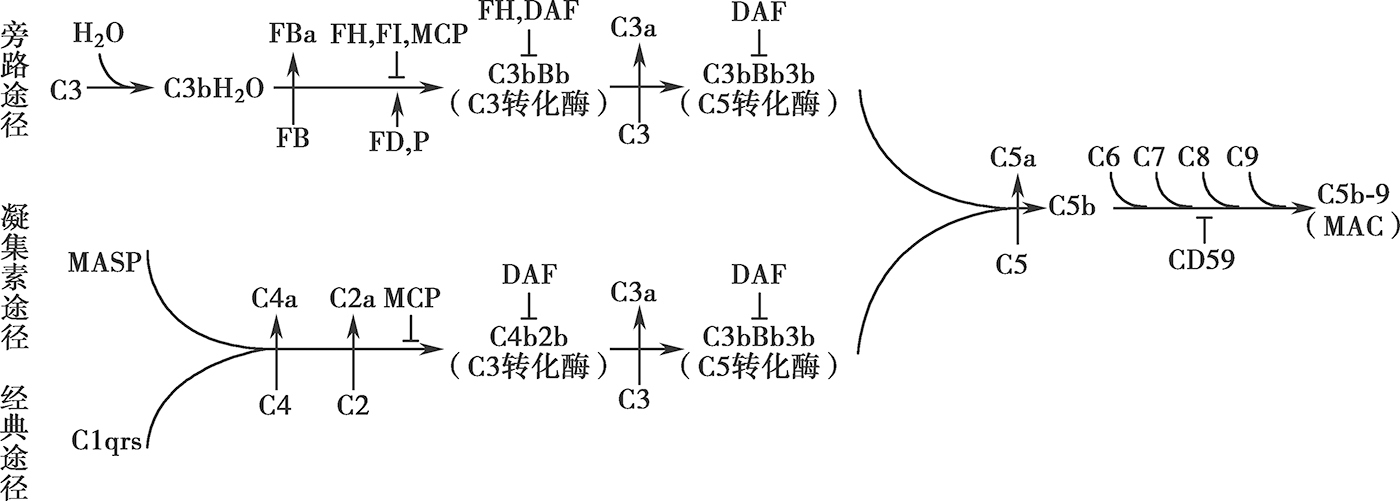
图6-2-1 补体系统的活化途径
注:MASP,凝集素相关丝氨酸蛋白酶;FB,B因子;FH,H因子;FI,I因子;MCP,膜辅因子蛋白;DAF,衰变加速因子;MAC,膜攻击复合物。
在补体系统中,多种调节蛋白发挥重要作用。如备解素促进C3b和FB之间的结合,从而稳定旁路途径C3转化酶。补体因子H和补体因子I(complement factor I,FI)紧密负调节溶液和自身细胞膜上的替代途径,CD59仅在自身细胞膜上调节旁路途径。
补体激活作为一把“双刃剑”,在宿主防御中发挥不可或缺的作用,但同时也能介导免疫损伤。经典的多重打击模型认为(图6-2-2),异常的半乳糖缺乏IgA1(galactose-deficient IgA1,Gd-IgA1)水平增加导致第一重打击。增加的Gd-IgA1引起自免疫反应导致抗聚糖抗体的产生,此为第二重打击。增加的Gd-IgA1及抗聚糖抗体形成免疫复合物沉积于肾小球系膜区,此为第三重打击。沉积下来的免疫复合物激活补体途径、刺激系膜细胞增生、导致细胞因子、趋化因子及细胞外基质蛋白释放增加,从而导致炎症及纤维化,这是第四重打击。而系膜细胞在补体激活中起主要作用,主要通过旁路和凝集素途径激活补体系统。
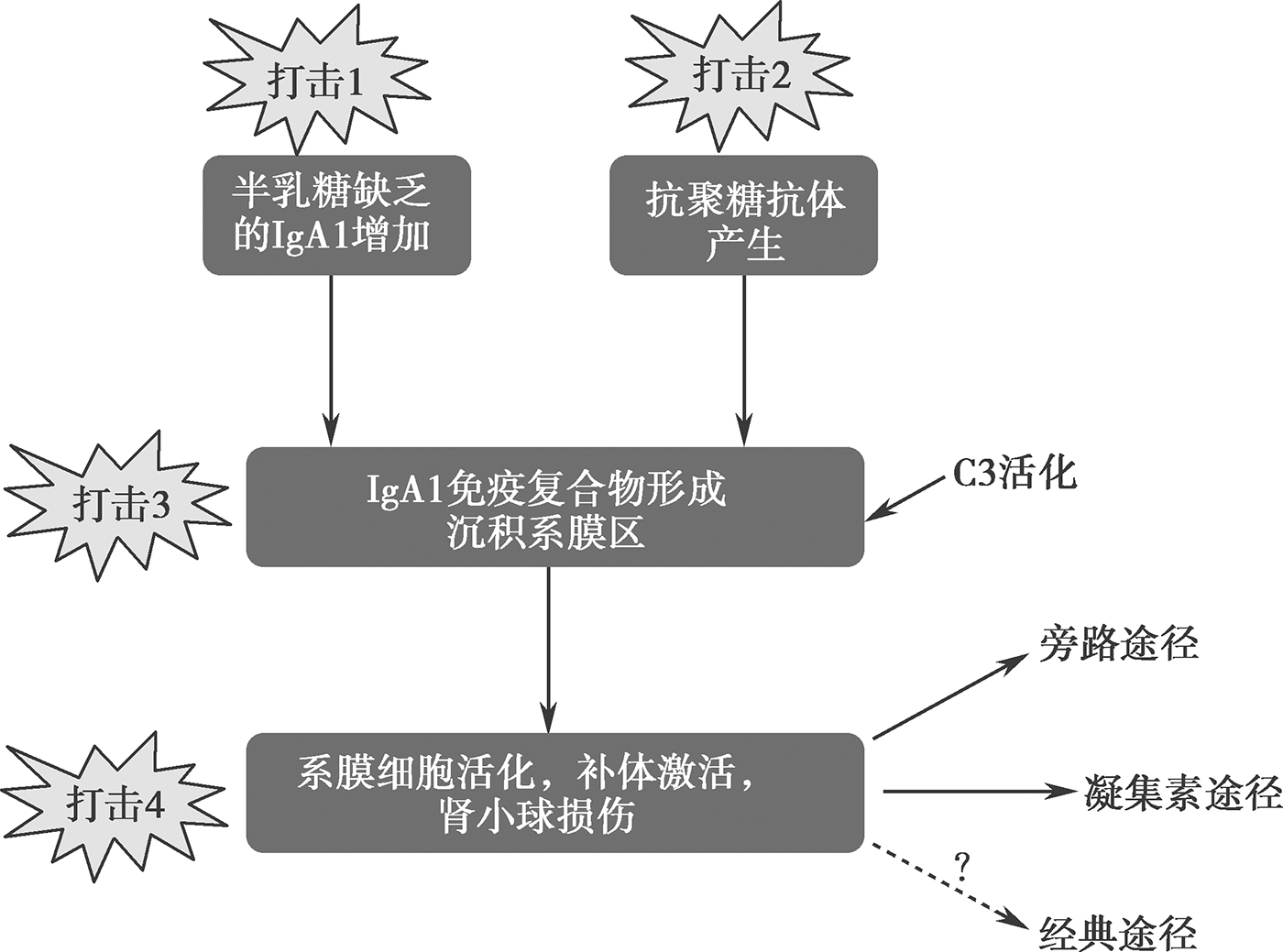
图6-2-2 IgA肾病多重打击模型
在一项无症状IgA肾病的尸检分析中发现,这部分人群系膜区并无C3的沉积。而已有多项研究表明,IgA肾病患者血C3水平的下降及系膜区C3的沉积强度,血MBL的缺乏及MBL在系膜区的沉积,均与不良的预后有关。说明在疾病进展期,补体旁路途径及凝集素途径被激活,C3及MBL被消耗较多。这些研究提示,补体系统被激活指向IgA肾病的进展。
旁路途径是IgA肾病中补体激活的主要方式。在75%以上的患者有发现备解素等对旁路途经有正向调节作用的成分与IgA、C3在肾脏共沉积。IgA单体不能激活补体反应,但多聚IgA(polymeric IgA,pIgA)及去糖基化的IgA1能直接激活旁路途径,但机制不明确。从全基因组关联研究(genome wide association study,GWAS)的研究结果来看,补体H因子相关蛋白(complement factor H related protein,CFHR)1和3基因的缺失可对IgA肾病有保护作用。
补体因子H(FH)是补体旁路途径的一个重要调节因子。FH分子的每个末端都呈现C3b结合位点。N末端C3b结合位点介导旁路途径C3转化酶(C3bBb)的加速衰变,以及介导FI依赖的C3b蛋白水解失活的辅因子活性。C-末端区域结合通常存在于细胞表面上的C3b和聚阴离子(例如,硫酸乙酰肝素和糖胺聚糖)。该区域对于FH在细胞表面上的补体调节活性以及区分自身和病原体至关重要,因为大多数病原体在其表面缺乏这些聚阴离子。CFHR基因位于编码FH基因的下游,由5个基因(CFHR1-CFHR5)组成,串联在1号染色体q32的补体活性簇调节子。CFHR蛋白与FH具有高度序列同源性,有短共有重复(short consensus repeat,SCR)框架。其C端SCR序列高度保守,对应于FH特定SCR区域,可识别C3b和细胞表面的部位。因此,五种CFHR蛋白中的每一种都与C3b和C3d结合并区分细胞和非细胞表面。CFHR蛋白可与FH竞争性结合C3b,导致FH失去对补体激活过程的控制。C3转化酶激活更多C3形成C3a沉积于肾脏组织,从而使组织出现损伤。有研究通过GWAS研究鉴定出,在CFH位点内 CFHR3 和 CFHR1 的基因突变,在IgA肾病、C3肾病中具有保护意义。而这种保护作用与较高的血浆H因子水平、较低的C3a水平及较少的系膜C3沉积有关。在分析20 612例患者的荟萃分析中发现,单核苷酸多态性(single nucleotide polymorphism,SNP)rs6677604(Chr.1q32)位点的突变具有显著的保护作用。单个等位基因的缺失可减少26%的IgA肾病患病概率,两个等位基因均缺失可减少45%的患病概率。但这种等位基因的缺失有很大的人种差别,如非洲裔有55%,而南美及东亚人群只有0~5%的缺失。在一项中国人群的对照研究( n =3 581),筛查有84-kb缺失的 CFHR3 和 CFHR1 基因。该项研究发现4.4%的IgA肾病患者和7.1%的对照人群有这2个基因的缺失。同时结果提示该缺失明显减少IgA肾病的发病风险,且使用牛津分型标准对临床和组织病理学参数分析发现,该缺失与肾小管间质损伤减少有关。但近期一项白种人的研究则发现虽然在 CFHR3 和 CFHR1 的基因缺失(无论是单个,还是2个缺失)人群中肾活检IgA、IgG、C3等的沉积强度较无基因缺失的人群弱,但该缺失与肾脏进展至慢性肾脏病3~5期无关。除了这两个基因编码的蛋白之外,CFHR5也被发现在少数IgA肾病患者肾脏中有沉积。另一项中国人群的研究发现与正常人相比,IgA肾病患者(N=1 126)外周血CFHR5水平显著升高(中位数4.55 vs 3.19μg/ml)。且高水平的血CFHR5与eGFR、高血压及IgA肾病牛津分型(T和C)有关,可作为IgA肾病进展的独立危险因素。有一项英国的研究发现,IgA肾病患者外周血CFHR5与更严重的IgA肾病牛津分型有关。
近年来有越来越多的证据支持凝集素途径在IgA肾病发病机制中的作用。从IgA肾病患者血清中提取的pIgA可激活凝集素途径。Endo等首次发现25%的IgA肾病患者有凝集素(MBL)/凝集素相关丝氨酸蛋白酶(mannan-binding lectin-associated serine,MASP)的沉积,而正常人群没有。且MBL沉积的频率高于其他的肾小球肾炎。有研究分析,发现MBL/MASP沉积的IgA肾病患者,其年龄较轻,肾穿刺前病程更短,且肾穿刺提示严重的肾脏损害,如系膜增生、新月体形成、小球硬化和间质纤维化等。有研究认为分泌型IgA有可能是MBL的配体。前者重链的N-聚糖有N-乙酰葡糖胺和甘露糖残基,有可能被MBL和纤维凝胶蛋白识别,从而启动凝集素途径。
由于基因编码多态性(多见于外显子1的第54个密码子),在健康人群中10%~20%存在MBL的缺乏。有研究比较了MBL缺乏和MBL充足IgA肾病患者的临床特征,发现前者的尿蛋白和eGFR水平均好于后者。Gong等通过分析MBL第54个密码子基因多态性与感染的关系后发现,携带突变等位基因( GAC )的IgA肾病患者,在起病或疾病恶化前呼吸道或肠道感染的事件多于野生纯合子(GGC/GGC)。近期有一项国内的回顾性研究(IgA肾病患者749例,正常人489例)检测了MBL2基因突变和外周血MBL水平,结果发现5.2%的患者有MBL缺乏(< 100ng/ml),其中LYPB/LYPB和LXPA/LYPB是主要的MBL2单倍型(32/39)。且MBL缺乏似乎与更高的前驱感染发生率和肉眼血尿的出现有关。另外,MBL缺乏与不良预后有关。
由于IgA肾病肾活检组织中C1q沉积极少,且C1q的出现一般与临床预后不良有关。因此,可认为在IgA肾病发病机制中无经典激活途径的参与,但与肾脏明显损害有关。另外,在肾小球中发现C4、C4d及C4结合蛋白的沉积,以往认为可能是经典途径激活的早期成分,但从目前的研究来看,更可能是凝集素途径参与的表现。