
下载掌阅APP,畅读海量书库
立即打开



无论从基因型还是从表型看,IgA肾病的发病及其肾衰竭进程均与遗传因素有关。详细内容参阅本书第二章相关内容。
IgA1分子糖基化异常与遗传因素也有关。例如在一项对同卵(27对)和异卵双生(47对)的健康女性孪生子中进行IgA1糖基化水平检测,发现Gd-IgA水平在同卵双生子中的相关性为0.84,而在异卵双生子中仅为0.46。
表观遗传调节在IgA分子糖基化异常中也发挥了重要作用。与健康对照相比,IgA肾病患者外周血单个核细胞中microRNA 148b、microRNA 188-5p、microRNA 361-3p、microRNA 886-3p、microRNA let-7b、microRNA let-7d表达上调,其中microRNA 148b可直接抑制C1GALT1的mRNA和蛋白表达,导致Gd-IgA1产生增多。
在IgA肾病患者肾活检组织中检测到microRNA 320表达上调,且与尿中的microRNA 320表达正相关。对患者B细胞进行检测发现,患者B细胞中的microRNA 320表达上调,直接作用于其靶基因PTEN,降低了 Cosmc 基因表达,刺激了B细胞增殖。
IgA肾病的发病常与黏膜感染有关。其依据是:①IgA肾病发病之前或发病的同时伴有呼吸道或胃肠道感染,这些感染与血尿关系密切,尤其是咽部感染。有学者称之为“咽炎同步性血尿”。由此认为系膜区沉积的IgA来源于黏膜分泌系统。②许多胃肠道黏膜病变易继发IgA肾病。③在一些IgA肾病患者的肾活检中发现麦胶、胶原蛋白、呼吸道病毒和肠道菌丛等成分及抗体。④血尿的产生与多聚IgA1产生有关。但随着研究的深入,发现黏膜浆细胞分泌的多聚IgA由2个单体、1个分泌片和1个J链构成。IgA肾病的系膜区IgA无分泌成分,仅有2个单体1个J链,这对IgA是否来源于黏膜系统提出质疑。有学者提出“黏膜-骨髓轴”说法,认为血清异常升高的IgA并非由黏膜产生,而是由黏膜内抗原特定的淋巴细胞或抗原呈递细胞进入骨髓腔,引起骨髓B细胞分泌IgA增加。
血清IgA1铰链区O-糖基化缺陷可能由于扁桃体B淋巴细胞缺陷所致。在日本IgA肾病感染副流感嗜血杆菌患者血清的抗IgA抗体滴度增加。用副流感病毒感染小鼠可以制作出IgA肾病模型。IgA肾病患者发生上呼吸道感染后可导致肾脏损伤加重。链球菌M蛋白刺激淋巴细胞产生TGF-β,而TGF-β可以增加血浆中产生IgA的细胞的数量。多种病毒感染都与IgA肾病的恶化有关,其中包括副病毒Ⅵ。一般来说,微生物感染可引起体内表达高水平的干扰素如(interferon-alpha,IFN-α),因而需要考虑阻断病毒感染后的IFN-α信号转导途径。对扁桃体的高反应性曾经被认为是IgA肾病的病因,但关于其免疫机制仍然存在不清楚的因素,如扁桃体炎是如何刺激骨髓产生IgA抗体的?脾脏中的外周B淋巴细胞参与对多糖抗原的反应,脾脏是否也在IgA肾病的发病中发挥作用?上呼吸道感染,比如扁桃体感染是否会影响肺部和支气管腺体的黏膜反应?
胃肠道和肝脏疾病的多种病理生理机制都和IgA肾病有关。毫无疑问,咽炎引起的血尿通常会与上呼吸道感染如支气管炎等有关。内在的炎症介质(如IFN-α)短时过量产生可以解释微生物可激发IgA肾病恶化的现象。IgA肾病有多种表型,但还没有研究提示IgA肾病的表型与哪些因素有关。细菌和病毒感染导致的IFN-α的波动会继发更加持久的IL-6的生成。有关免疫细胞区室化的知识还知之甚少,但情况正在改变,随着研究的深入,是有可能解释食物中的抗原是如何加重IgA肾病的。
所有的细胞都可以产生干扰素-γ(interferon-γ,IFN-γ)。分泌的IFN-γ有以下功能:刺激NK细胞增生,引起感染细胞的细胞毒性;激活Th1辅助细胞和毒性淋巴细胞;激活γδT细胞并影响抗体产生;影响树突细胞的功能;促进其自身的自分泌。口服IFN-γ可产生类似于黏膜感染引起的反应而非系统感染引起的反应。黏膜趋化因子(C-C motif chemokine ligand 28,CCL28)通过其受体(C-C motif chemokine receptor 10,CCR10)选择性吸引浆母细胞,在IgA肾病患者的肾小球内,IFN-γ促进Th1淋巴细胞的免疫反应。中度水平的IFNα可以增加IL-12对Th1淋巴细胞反应,但是高水平的IFN-α会抑制IL-12的生成和NK细胞产生IFN-γ。在儿童IgA肾病患者中,IFN-γ可以促进CD8淋巴细胞在肾小球的募集。IFN-γ还可以促进CD4细胞表达CD40配体。扁桃体中有CD4 + T细胞和B细胞,还有可以产生TNF-α的单核细胞,这些细胞都可以迁移到血液。扁桃体中有的B淋巴细胞具有异质性,多数B淋巴细胞只产生IgM,但其他细胞可以像脾脏边缘区B细胞一样产生IgM和IgD,只有少数可以进入血液。
还有一些炎症因子也在IgA1分子糖基化异常中发挥了重要作用。例如IgA肾病患者体内APRIL水平升高,且与半乳糖缺陷的IgA1水平和蛋白尿严重程度正相关,与eGFR负相关。体外培养患者的B淋巴细胞,予APRIL刺激,可较正常人产生更多半乳糖缺陷的IgA1。IgA肾病患者体内a-防御素中性粒细胞肽水平较正常人明显升高,且与Gd-IgA水平负相关,而与肾功能及中性粒细胞计数无关。TGF-β、IL-17和IL-4可以通过抑制 C1GalT1 和 Cosmc 基因表达,导致DAKIKI细胞分泌糖基化异常的IgA1。分离IgA肾病和健康对照人群血及扁桃体中分泌IgA1的B淋巴细胞,EB病毒转染形成永生细胞系后,进行体外培养,加入IL-6刺激,通过上调及延长信号传导、转录激活蛋白3(signal transducer and activator of transcription 3,STAT3)磷酸化,导致该淋巴细胞分泌Gd-IgA1增多,使用STAT3的抑制剂,可减少IL-6诱导的磷酸化,降低Gd-IgA1分泌,JAK抑制剂也有类似效果,使用Kinomic分析发现,IL-6通过JAT/STAT3/MAPK信号通路调节Gd-IgA1分泌。
免疫球蛋白大多为糖蛋白,其寡糖链对维持蛋白分子的结构和功能有着极其重要的作用,如糖链的改变会明显影响整个分子的代谢,改变其与各种受体的结合力,降低对致病细菌的调理能力及对细菌黏附作用的抑制,影响激活补体的能力。近年发现免疫球蛋白糖链结构的异常与一些自身免疫性疾病如IgA肾病、类风湿关节炎等的致病机制密切相关。IgA肾病患者IgA1糖基化异常上文已详细叙述,此处不再赘述。
合成IgA1分子铰链区的氨基酸,利用N-乙酰氨基半乳糖苷转移酶添加GalNAc,在2,3-唾液酸转移酶和2,6-唾液酸转移酶作用下GalNAc上增加唾液酸。糖复合物中糖链的合成没有模板,而是通过一系列定位有序的糖基转移酶来完成的,因此,在糖生物学中提出了一个基因→一种转移酶→一个连接键的扩展中心法则。IgA1铰链区O-糖链末端的Gal是以β1,3糖苷键与GalNAc连接的,此糖苷键是由β1,3-半乳糖基转移酶(β1,3-GT)催化形成的。β1,3-GT是功能各异但具有同源基因的酶家族,现已确认的有10种同源基因表达的不同酶,它们作用于不同供体(UDP-Gal和UDP-GlcNAc)与不同的糖受体(GlcNAc、Gal、GalNAc),最终都形成β1,3糖苷键。当然也有研究认为,单纯IgA1分子糖基化异常并不是造成IgA肾病的原因,更可能与患者体内B细胞糖基合成酶的缺陷有关,因为IgA肾病患者体内的IgA1分子的GalNAc含量与正常人相似,提示可能患者体内有2种产生IgA1的B细胞,由于其半乳糖转移酶对IgA1铰链区GalNAc利用的差异,一种产生正常糖基化的IgA1分子,另一种产生富含Tn抗原的IgA1分子。IgA肾病患者IgA1的O-糖链末端的半乳糖缺失可能由于β1,3-GT的表达降低或酶的结构异常而导致酶活性下降。更详尽的研究排除了 β1 , 3 - GT 基因缺失的因素,证明是酶本身活性的降低。体外对β1,3-GT活性丧失的细胞进行处理(用去甲基试剂如5-氮胞苷)后可使β1,3-GT的酶活性恢复,能催化产生正常糖基化的O-糖链。为了确定IgA肾病中β1,3-GT的活性改变,将正常IgA1铰链区O-糖链用β-半乳糖苷酶去掉半乳糖基后得到半乳糖基受体,分别与来自IgA肾病患者和正常人的T、B细胞和单核细胞裂解液共培养,使之重新半乳糖基化。然后用生物素化的凝集素VV(Vicia Villosa,对 GalNAc 特异)测定受体的半乳糖基化程度,结果与T细胞和单核细胞共培养的β1,3-GT活性在IgA肾病与正常人间无差异,而对于与B细胞共培养的β1,3-GT活性,IgA肾病患者的显著低于正常人,并且其酶活性与VV和IgA的结合率负相关,从而提出B细胞的β1,3-GT活性低下与IgA肾病的发病有关。体外培养扁桃体单个核细胞发现,IgA肾病患者基线BAFF、Gd-IgA1水平显著高于慢性扁桃体炎不伴有肾损害患者,而C1GALT1和Cosmc mRNA表达显著低于后者,对培养细胞进行不同频率震荡后,这种差异更加显著。患者体内Cosmc水平显著低于正常人。用LPS刺激患者外周血B细胞,导致低糖基化IgA的分泌增加,注射北黄芪提取物可以显著增加Cosmc活性,减少IgA分泌,剂量依赖性逆转糖基化降低的水平。辣椒素也可以诱导IgA肾病患者扁桃体单个核细胞中BAFF的表达,抑制C1GALT1和Cosmc表达,导致低糖基化IgA1的表达增多。
表观遗传调节在IgA肾病糖基转移酶活性调节中也发挥了重要作用。人体不同组织的Gal转移酶和 Cosmc 启动子区是相似的,由于不同的DNA甲基化造成了表达量的差异,这可能是患者和正常人群IgA1分子半乳糖基化存在差异的原因。IgA肾病患者体内分离出的B细胞中let-7b表达显著高于非IgA肾病患者及健康对照,let-7b可以下调人体外周血单个核细胞中GALNAc转移酶2的表达。IgA肾病患者外周血B淋巴细胞Cosmc mRNA表达显著低于其他肾炎患者及健康对照人群。这些B细胞体外培养后,使用IL-4处理,发现 Cosmc 启动子区DNA甲基化在IgA肾病患者显著升高,而DNA甲基化抑制剂5-AZA可显著降低IgA肾病患者 Cosmc 甲基化水平。 Cosmc 启动子区DNA甲基化水平与 Cosmc 基因mRNA表达负相关,与培养液中半乳糖缺失IgA1水平正相关。
分离患者和健康对照人群循环中分泌IgA1的细胞,评估细胞因子能否改变IgA1糖基化,结果显示IL-6和IL-4可以显著升高IgA1半乳糖缺陷的程度,在IgA肾病患者体内改变更明显,这些细胞因子可以直接降低C1GalT1表达,间接升高ST6 N-Acetylgalactosaminide Alpha-2(ST6 N-乙酰氨基半乳糖α-2)、6-Sialyltransferase Ⅱ(6-唾液酸转移酶Ⅱ)、ST6 Gal-NAc-Ⅱ(ST6 Gal-N-乙酰氨基半乳糖α-2)的表达,提示IL-6和IL-4通过调节关键酶活性增加了半乳糖缺失。IgA肾病患者外周血B细胞的ST6 Gal-NAc-Ⅱ表达较正常对照显著降低,且与患者唾液酸缺陷水平相关。
在IgA的代谢和IgA1-IC的清除过程中有2个主要受体参与:①去唾液酸糖蛋白受体(ASGPR),在肝细胞上表达,通过识别绞链区的O-糖链而与IgA结合;②IgAFc受体(FcαR),在中性粒细胞、单核细胞和嗜酸性粒细胞等有表达,能够与单体、二聚体IgA1和IgA-免疫复合物结合。将放射性标记IgA1注入IgA肾病患者体内,结果显示肝细胞对其清除正常,说明ASGPR在IgA肾病中没有异常。扫描分析显示IgA肾病患者的单核细胞表面FcαR的表达比正常人明显下降,SDS聚丙烯酰胺凝胶电泳法(SDS-PAGE电泳)发现FcαR的泳动速度稍高并且与唾液酸特异的凝集素的结合下降,说明IgA肾病的FcαR的异常可能是在翻译后发生的唾液酸化降低。
单核细胞在无血清培养时FcαR表达升高,与自体血清共培养时则明显下降,提示循环中存在调节因子;同时诸如艾滋病(acquired immune deficiency syndrome,AIDS)等伴有血清IgA水平升高的疾病中都有FcαR的表达下调,提示IgA在FcαR表达调控中起作用。正常人单核细胞与血清IgA(5mg/ml)长时间共培养,FcαR的表达持续下降,在IgA缺陷的患者中FcαR强表达,多聚IgA对FcαR的结合力比单体IgA强并使FcαR的表达下调更明显,因此IgA特别是多聚IgA或IgA-IC在单核细胞FcαR表达下降中起主要作用。
IgA肾病患者的血清中多聚体IgA和IgA-IC增加,因此导致FcαR表达下调。IgA肾病患者的IgA水平升高使IgA与FcαR的结合增加。其次,与正常单核细胞培养发现,IgA肾病患者的IgA与FcαR的结合力比正常人IgA(IgA浓度相同)要高1~2倍,说明IgA肾病中异常糖基化的IgA可能对FcαR的占据起直接作用。FcαR与IgA的结合位点在其铰链Cα2和Cα3交界处,Cα2区的糖基化可能对FcαR与IgA相互作用起关键作用。另外,去唾液酸化的骨髓瘤IgA与FcαR的结合力增强,用唾液酸酶处理去除FcαR上的唾液酸,结果IgA与FcαR的结合增加5倍,证明了IgA肾病中FcαR的唾液酸化异常所引起的受体与配体间的亲和力增强是IgA1与FcαR结合力增强的另一可能原因。因此在FcαR数目减少的情况下,IgA与FcαR的结合仍增加。IgA肾病患者单核细胞表面的FcαR被IgA持续、高度占据,致使胞吞动力下降,受体上结合的IgA可再次进入循环,这使IgA和IgA-IC免受胞内溶酶体的降解,而增加了它们在血清中的浓度,可能对肾脏系膜细胞IgA的沉积起到间接作用。此外,与FcαR结合的IgA与抗原、免疫复合物和/或肾脏系膜细胞的IgA受体结合,释放出炎性介质肿瘤坏死因子-α(TNF-α)和IL-6,TNF-α引起细胞凋亡而直接损伤肾脏系膜细胞,而IL-6促使肾脏系膜细胞增生,因此FcαR和IgA的异常作用对IgA肾病的致病有直接作用。
FcαR在肾脏系膜细胞无表达,但系膜细胞既可以结合单体IgA,又可以结合聚体的IgA,这提示系膜细胞上存在IgA的另一种受体。实验证明人和鼠的系膜细胞上至少有1种IgA受体(IgA-R)。 131 I-IgA结合到系膜细胞上随剂量的增大而增大并在1.5mg/ml时达到饱和,未标记的IgA及其Fc片段对结合有抑制作用,而IgG、IgM和IgA的Fab片段没有,说明此受体是IgA特异性的。去唾液酸IgA和IgA分别与系膜细胞结合达到饱和时,去唾液酸IgA结合的数量较多。对于分别加galactosamine(Gal)、N-Acetyl-D-galactosamine(GalNAc)和N-Acetylglucosamine(GlcNAc)培养的系膜细胞,IgA与受体的结合明显下降,其中GalNAc的抑制效果最明显,同样这些糖对去唾液酸IgA的抑制要比对IgA的抑制强得多,这表明IgA分子中的铰链区的O-糖链在IgA与系膜细胞上受体结合中起主要作用,这一结果也进一步表明IgA1的O-糖链的异常在IgA肾病致病机制中的重要作用。有学者在人系膜细胞上找到了识别IgAFc段的受体,并证明与单核细胞的FcαR不同,但可能具有部分同源性,此受体对多聚体IgA的结合力大于单体,这为大量多聚体IgA在肾脏系膜细胞上沉积提供了依据。
免疫功能异常是IgA肾病的主要致病因素。35%~50%的IgA肾病患者血清中IgA含量升高,其中,主要是聚合型的IgA1亚型,并常以大分子的免疫复合物(IgA-IC)形式存在。研究显示IgA水平的升高与T细胞和B细胞的作用有关。IgA肾病患者可产生IgA1的浆细胞数量增加,并且所产生的绝大多数是多聚IgA1,目前尚未找到其特异的抗原,因而认为多聚IgA可能是不依赖抗原的“天然”抗体,由多克隆活性B细胞生成。IgA肾病患者外周血中的B细胞,在无刺激物或有丝分裂原刺激下都能分泌异常高水平的IgA,其表面IgA的表达增加。B细胞的IgA分泌尚受到T细胞的调控,IgA肾病患者外周血单个核细胞中T细胞数量增加,经实验证明主要是Tγ和Tδ细胞的数量增加,并且与表面有IgA表达的B细胞数成比例。将IgA肾病患者的外周血细胞在体外经有丝分裂原刺激,其IgA合成水平显著升高,去除Tγ和Tδ细胞则无此现象。从IgA肾病患者外周血单个核细胞纯化的Tγ和Tδ细胞比Tα和Tβ细胞能诱导B细胞产生更多的IgA分子,并且激活后的Tγ和Tδ细胞能产生大量的转化生长因子-β(TGF-β),它是启动B细胞从IgM合成向IgA合成的主要细胞因子之一。因此,IgA肾病患者Tγ和Tδ细胞的增加,增强了B细胞对IgA的合成。
IgA肾病患者血清IgA的变化是由干细胞决定的,其主要依据是:①将有IgA肾病倾向的ddY鼠的骨髓细胞移植到B6鼠后,增加了受者血清中的大分子IgA和肾小球上IgA的沉积;②对1例IgA肾病合并慢性粒细胞白血病患者进行同种异体骨髓移植后,不仅治愈了白血病,而且肾脏系膜细胞上沉积的IgA也被清除了。进一步实验发现,约5周龄的产生高浓度IgA的鼠移植了正常B6鼠同种异体骨髓后,肾脏系膜细胞的IgA和C3补体的沉积减少,肾小球硬化和肾系膜基质增生程度减轻,该研究提示:同种异体骨髓移植可能是治疗IgA肾病的一种新手段。目前,骨髓移植治疗自身免疫疾病正受到瞩目。单纯IgA水平的增高并不能解释肾脏系膜细胞上IgA的沉积,因为骨髓瘤和其他一些可引起IgA增高的疾病,几乎不伴有IgA免疫球蛋白在肾组织沉积。因此,推测在IgA肾病中,IgA分子存在着免疫学或化学结构的特征性变化。
虽然补体的级联反应不是IgA肾病发展的必需因素,但有证据表明局部补体的活化可以影响肾小球损伤的程度。研究表明,大鼠的二聚体和多聚体IgA能够通过旁路途径激活补体而造成肾小球损伤,单体则没有这种效应。系膜区IgA对C3的活化可能是通过MBL途径发生的,最终产生C5b-9,但其浓度不足以引起系膜细胞溶解,在此基础上进而活化系膜细胞产生炎症介质和ECM。C3和MBL不仅可以在肾脏沉积,还可以在局部由系膜细胞合成。此外,C3还可以由足细胞合成。因此,系膜细胞一旦结合IgA,可能利用自身产生的C3和MBL在局部活化补体,而不依赖于系统补体的活性。这种原位的补体合成和活化在肾小球损伤进展中的作用尚不清楚。系膜细胞还可以合成补体调节蛋白,如DAF,这是IgA肾病患者中产生的C5b-9通常不导致系膜细胞溶解的可能原因。IgA肾病时也明显存在着通过旁路途径而激活补体的证据。组织学检查发现,在系膜C3沉积时,缺乏经典途径和MBL途径的早期成分(C1、C2或C4),这对补体旁路途径活化在IgA肾病的发病机制中发挥作用是一个有力证据。
补体C3的受体CR1(CD35、C3b受体)在大多数循环中的细胞(红细胞、单核细胞、B细胞、多形核白细胞等)及肾脏足细胞表面都有表达。而CR2(CD21,C3dg的受体)仅表达于B细胞及树突状细胞。CR1是人类肾脏表达的唯一C3受体。在IgA肾病或紫癜性肾炎患者肾活检标本检测到CR1的表达与正常肾脏没有区别。因为系膜细胞不表达C3受体,因此很明显C3受体在IgA肾病中没有参与IgA-C3免疫复合物的肾沉积。另一方面,如果足细胞内的CR1表达下调,可以使足细胞对补体攻击的敏感性增高。
系膜细胞可以产生C3及补体调节蛋白——衰变加速因子(decay accelerate factor,DAF)。正常状态下,DAF是由球旁器产生的,而且生成很少,几乎检测不到。但在疾病状态下,系膜细胞、小管细胞和炎症细胞都能合成DAF和C3。在IgA肾病,系膜细胞DAF和C3的阳性率和系膜炎症及小球硬化程度正相关。此外,小球细胞C3-DAF的转化率和小球损伤程度相关。用免疫复合物刺激系膜细胞可以通过其Fc受体诱导C3的合成。C3的生成可以进一步调节细胞因子如IL-1的产生。
在IgA肾病过程中,含IgA的免疫复合物沉积的基础上,局部补体合成及随后的补体活化可造成组织损伤。补体的生成可以被免疫复合物和肾小球固有细胞及炎症细胞产生的细胞因子诱导和调节。
来自患者和动物模型的大量资料表明,IgA肾病的发生是多种致病因子共同作用,从而促进病情发生、发展的一个过程。其必须的3个阶段包括:①异常大分子IgA1的形成及其对可溶性FcαRⅠ的屏蔽,并通过跨膜FcRγ相关性的FcαRⅠ信号传递激活单核细胞;②通过大分子IgA1和系膜IgA受体的相互作用导致系膜细胞激活;③通过数种炎症激活途径导致系膜及白细胞的激活,共同促进病情的发展。
使用Gd-IgA1和链球菌M4蛋白与人肾小球系膜细胞共孵育,可导致系膜细胞显著增生,分泌更多的IL-6、血小板源生长因子(platelet-derived growth factor,PDGF)和C3。从IgA肾病患者体内分离出IgA免疫复合物,分子量800~900 000,包含的IgA1分子存在明显半乳糖缺陷,刺激人系膜细胞后,通过增加苏氨酸磷酸化诱导系膜细胞明显增生,分泌更多IL-6和IL-8。对系膜细胞刺激作用最明显的患者快速进展至终末期肾病(end-stage renal disease,ESRD)。而在体外制备半乳糖和唾液酸缺陷的IgA1,与正常人和IgA肾病患者血清混合,通过蛋白分离柱提取IgA1-IgG免疫复合物,与系膜细胞共孵育,发现和健康对照人群相比,患者提取出的免疫复合物可通过脾脏酪氨酸激酶信号通路,导致MCP-1、IL-6、IL-8、IFN-γ诱导蛋白10、血小板生长因子-BB等表达增高,增加Ⅰ型组蛋白去乙酰化酶表达,明显诱导系膜细胞增生,分泌更多细胞外基质。
IgA沉积到系膜细胞后,会通过系膜细胞-足细胞对话导致足细胞损伤。研究发现,与健康对照人群相比,IgA肾病患者血清提取的IgA分子热聚合后刺激小鼠系膜细胞获得上清液与足细胞共培养,通过活化MAPK/ERK通路,导致足细胞自噬减少,凋亡增加,nephrin表达下降,细胞骨架重组,血小板活化因子、TNF-α、TGF-β等分泌增加,肾素、血管紧张素、血管紧张素转换酶mRNA表达明显升高,依那普利、缬沙坦或糜蛋白酶抑制剂可增加nephrin表达,降低血管紧张素及其转换酶的高表达,改善足细胞的功能。
综上所述,IgA肾病的发病分子机制甚为复杂。本章总结其可能的机制为:可能存在的遗传背景因素、表观遗传调节、上呼吸道感染所产生的细胞因子、糖基化转移酶活性异常以及其他的未知因素导致的免疫球蛋白IgA的血清含量增加及其O-糖链的糖基化异常。IgA的O-糖链的糖基化异常,随后被抗糖基抗体IgG(或IgA1)识别,形成的免疫复合物太大,不能进入肝Disse腔,逃避了正常的清除机制,到达肾循环,通过了肾小球毛细血管孔径较大的内皮连接,沉积于系膜区,这些免疫复合物结合到系膜细胞上。高水平的IgA使其受体表达下降而使IgA代谢异常、IgA1的糖基化异常又增大了其与肾系膜上受体的结合力,以及抗多糖抗体的产生增加等,增加了系膜细胞-足细胞对话等一系列反应引起了肾小球的损伤(图3-3-1)。这些特点证明IgA肾病是一个自身免疫疾病,糖基化异常的IgA1是其自身抗原。
IgA1糖基化异常的根源可能与干细胞、基因的多态性密切相关,目前IgA肾病的遗传因素越来越成为人们关注的焦点,有关遗传因素的论述可参阅本书第二章的相关内容。
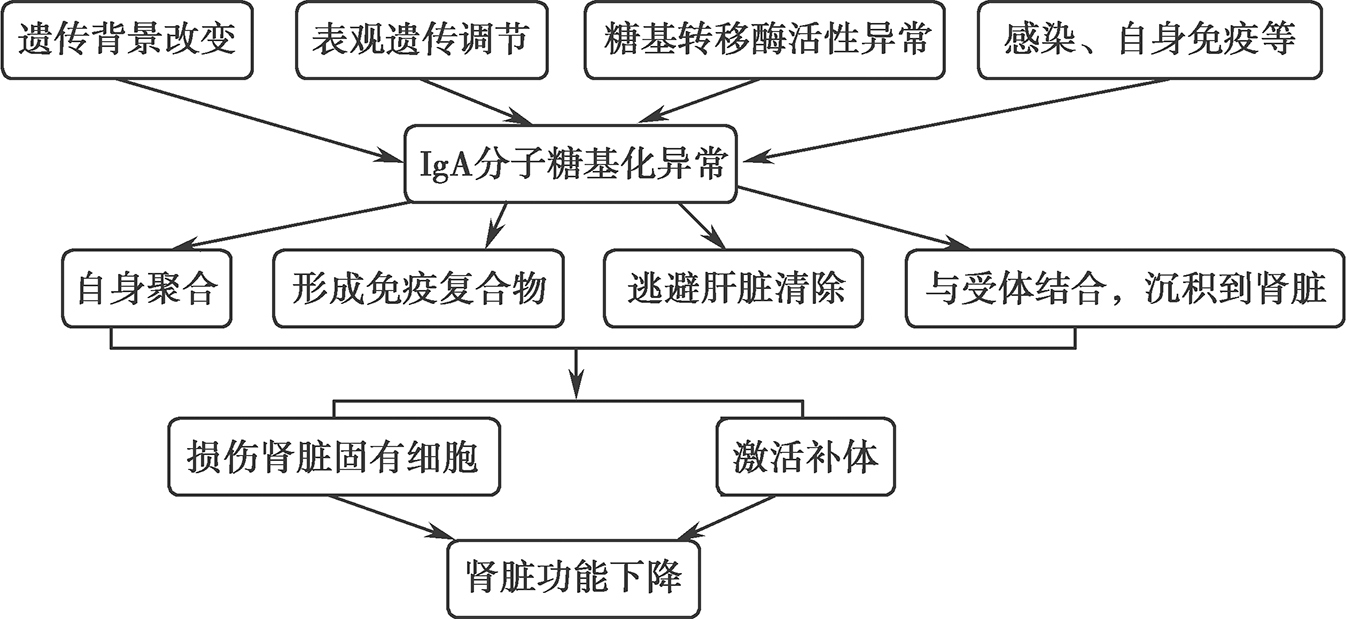
图3-3-1 IgA分子糖基化异常与肾小球的损伤
(徐丽霞)