
下载掌阅APP,畅读海量书库
立即打开



脓毒症(sepsis)是医学领域最古老、最难以捉摸的综合征之一,是当前医学界所面临的棘手难题。早在古希腊时期,希波克拉底将脓毒症定义为鲜肉腐败、沼泽产生污秽气体、伤口溃烂等过程。而后来的盖伦则认为脓毒症是有益的过程,为创伤愈合所必需。随着巴斯德等人的病原体理论的确立,脓毒症被定义为全身性的感染,认为是病原菌入侵机体血液系统并扩散的结果,经常被描述为“血液中毒”。抗生素应用的进步,可以完全清除脓毒症患者血液中的病原菌,但患者还是死亡。这使得病原体理论不能完全解释脓毒症的病理生理过程。随着人们对感染时机体病理生理变化过程的深入研究,人们发现,虽然脓毒症是由感染所引起,但其发展遵循着自身的病理过程规律:感染-脓毒症-器官功能障碍-死亡。因此,从本质上讲,是机体本身,而非病原体,介导了脓毒症的病理生理过程。
在脓毒症及多器官功能障碍综合征(MODS)的发生机制认识过程中,从机体本身的角度出发,人们提出了全身炎症反应综合征(systemic inflammatory response syndrome,SIRS)的概念。在临床实践中,临床医生在发现创伤、感染、胰腺炎等多种情况下,患者往往出现心率加快、呼吸频率加快、发热、白细胞计数增加等一系列的临床表现。与此同时,随着免疫、生物学技术等方面的发展,在基础研究中发现了一系列与脓毒症和器官功能障碍相关的细胞因子(如TNF-α、IL-6、IL-8等)。至此,炎性介质-SIRS-sepsis-MODS之间的关系日益清晰。为使研究更加标准化,建立起共同的交流平台,进一步明晰脓毒症相关概念,有助于临床上对脓毒症的早期诊断和治疗,并加深对脓毒症及其严重后果发生机制的理解。1991年,美国危重病医学会和胸科医师学会(ACCP/SCCM)联席会议,对SIRS、脓毒症等相关的概念作出了明确的定义。
SIRS指由各种感染和非感染因素作用于机体,所导致的一系列全身性炎症反应的过程。其病因包括两类情况:①由感染引发的SIRS,应更确切地称之为脓毒症。②非感染性病因:如出血性休克、缺血、多发性创伤、组织损伤、急性胰腺炎、中毒、烧伤及药物热等同样可以引发SIRS。
其诊断是具备以下两项或两项以上体征:①体温>38℃或<36℃;②心率>90次/分;③呼吸急促,频率>20/分,或过度通气,PaCO 2 <32mmHg;④白细胞>12×10 9 /L,白细胞<4×10 9 /L或幼稚细胞>10%,但应排除可以引起上述急性异常改变的其他原因(如化疗等)。
会议提出脓毒症(sepsis)是由感染引起的SIRS;增加了严重脓毒症(severe sepsis)的概念:脓毒症并发急性器官功能障碍;脓毒性休克(septic shock):脓毒症并发对液体复苏无反应性的低血压或者高乳酸血症。同时,会议摒弃了败血症(septicemia)的概念,提出了菌血症(bacteremia)的概念:血液内存在活菌,仅指在血培养阳性,而无明显临床症状,如果血液中存在病毒、真菌等则分别称之为病毒血症和真菌血症等。认为MODS是SIRS发展的最终结果。随着SIRS相关概念的明晰和基础研究的进展,使人们对脓毒症及其后果的发生机制有了更深刻的理解。为临床上最终解决这一棘手问题提供新的思路。
此次会议后,脓毒症相关的概念得到广泛的推广和应用,经过10年的应用,人们在应用过程中发现了一些新的问题。为了进一步改进相关内容,于是在2001年12月,即1991年的ACCP/SCCM共识会议10年后,美国危重病医学会(SCCM)、欧洲加强治疗医学会(ESICM)、美国胸科医师学会(ACCP)等5个学术团体,在华盛顿再次共同讨论了脓毒症的相关问题,最终形成的共识归纳为以下几点:
1.全身炎症反应综合征 认为可以由感染因素和非感染因素诱发,为强调非感染因素的作用,继续保持SIRS这个术语仍然是必要的。但也同时承认,1991年制订的SIRS诊断标准由于过度敏感和缺乏特异性而难以被临床使用。虽然全身炎症反应综合征的表现可以变化很大,但有共同的炎性介质和生化物质为基础。因此专家们希冀,未来诊断SIRS能够不再像当前依靠临床症状,而是使用生化或免疫学方面的指标。
2.感染 感染原被定义:病原性的或潜在病原性的微生物侵入正常时无菌的组织、体液或体腔的过程。但这个定义是有缺陷的,病原微生物造成感染所侵入的环境未必一定是无菌的,可以是有菌的;感染表现也未必一定是由微生物引起,可以由细菌毒素所致。对此最典型的例证是由难辨梭状芽孢杆菌毒素所导致的假膜性结肠炎。必须强调,临床上许多感染即使没有细菌学证据也不能排除,包括脓毒症在内,允许在缺乏细菌学证据的情况下高度予以怀疑。
3.脓毒症 仍维持1991的定义:感染引起的全身炎症反应。与对SIRS不同,为能够确保感染尽快得到有效控制,对脓毒症的诊断标准的要求是把敏感性放在首位。专家们认为,原SIRS诊断标准并不足以用来进行脓毒症诊断,因此,推荐了一系列机体对感染应答的全身表现,让医师能够较SIRS标准更准确地判断感染患者进入“脓毒症”状态(表15-1)。但要强调,这些征象仍然是非特异性的,因此,排除性的诊断十分重要;另外,表中提出的阈值也值得讨论。
表15-1 脓毒症诊断标准

a.定义为一个由微生物所引发的病理学过程;b.在儿童,>70%是正常的(正常值为75%~80%),因此,在新生儿和儿童不应被视为脓毒症的征象;c.在儿童,3.5~5.5是正常的,因此,在新生儿和儿童不应被视为脓毒症的征象;d.在婴幼儿,脓毒症的诊断标准是炎症反应的体征和症状再加上感染,并且伴有发热或低温(直肠温度>38.5℃或<35℃)、心动过速(在低温时可以缺乏)以及至少下列一项器官功能改变的提示:意识变化、低氧血症、高乳酸血症和跳跃的脉搏(bounding pulses)
4.严重脓毒症 定义为合并器官功能障碍的脓毒症。器官功能障碍可以通过Marshall评分系统(1995年)或序贯器官衰竭评估评分进行诊断(表15-2)。
5.脓毒性休克 亦称为感染性休克,指其他原因不可解释的,以低血压为特征的急性循环衰竭状态。其诊断标准:收缩压<90mmHg(在儿童<2SD)或收缩压减少>40mmHg;平均动脉压<60mmHg;毛细血管再充盈>2秒;四肢厥冷或皮肤花斑;尿量减少。
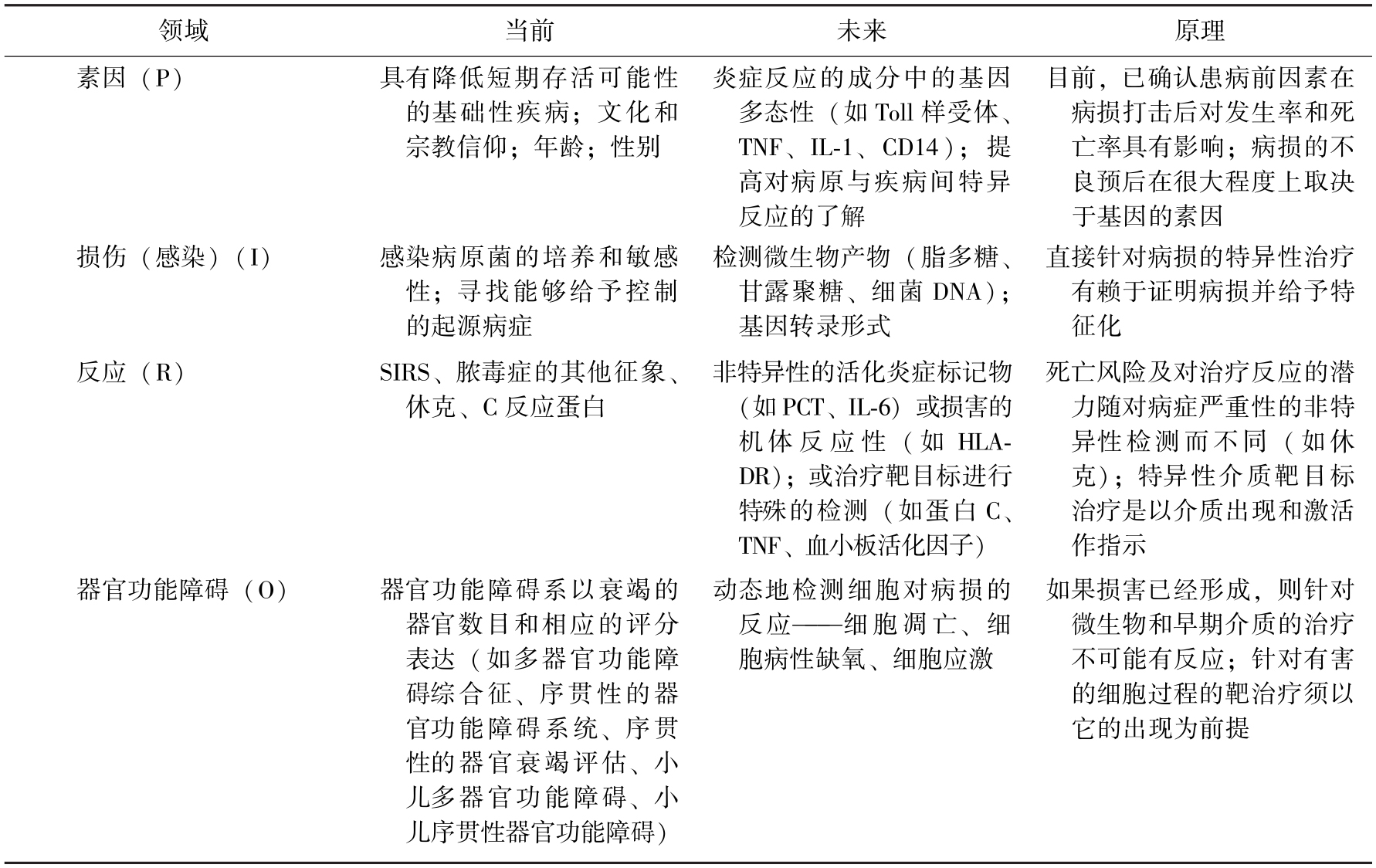
6.会议提出了一个脓毒症的分阶段系统(staging system) 会议认识到,以上术语的定义和诊断方法并不能保证对患者的准确诊断,它们只是基于当前条件下的阶段性产物,与会者希望最终能够发展出一个类似肿瘤患者诊断的TNM系统用于脓毒症等相关诊断。为达到上述目标,会议推荐了一个PIRO项目作为脓毒症诊断的“分阶段系统”,详细阐述了各项目的内容(表15-3)。
表15-2 严重脓毒症诊断标准

表15-3 阶段性的脓毒症诊断发展PIRO系统
能激活机体各种炎症介质大量释放的因素均可引发全身炎症反应综合征(SIRS),通常可分为感染性因素和非感染性因素。感染性因素引起的SIRS即脓毒症,感染在外科领域尤为常见,是引发SIRS的常见原因,感染源侵入机体以后,在机体内繁殖、释放毒素,引起机体组织变性、坏死,刺激机体的免疫应答反应,变性坏死的组织及激活的免疫活性细胞均可产生各种免疫炎性介质。若原发病能及时得到控制,有害刺激因素被清除,机体的损伤得以修复,炎症反应逐渐减低至可控范围,则疾病好转,SIRS消失;反之,SIRS的进一步发展,则可导致MODS、脓毒性休克甚至死亡。非感染性因素包括严重创伤、烧伤、胰腺炎、免疫系统疾病、缺血-再灌注损伤等,和感染性因素一样,同样可以激发机体的炎症瀑布式反应。通常很多非感染性因素导致的机体损伤,如果得不到及时有效的控制,后期并发感染的几率很大。因此,感染性因素和非感染性因素在SIRS的发生、发展过程中相互关联。
1.机体损伤时局部的炎症反应 炎症是具有血管系统的活体组织对损伤因子所发生的防御反应。任何损伤因子在引起细胞和组织损伤的同时,也激活机体产生免疫防御反应。机体与生俱有的自身防御能力通常基于三个方面:①皮肤、黏膜等外部屏障,阻止病原体的入侵和各种机械性损伤;②吞噬细胞、自然杀伤细胞等非特异性免疫系统清除外来入侵的病原体;③抗原特异性的高级免疫应答反应。血管反应是炎症过程的主要特征和中心环节,血管反应导致血管内的液体和白细胞渗出进入损伤部位,杀灭和清除有害因子。炎症反应的局部过程可以总结为以下4点:损伤后局部血流动力学改变,血管通透性增高,局部的液体渗出和白细胞游出。以外科感染为例,微生物通过局部损伤处侵入机体并繁殖,炎症反应被迅速激活、放大,并将损伤的信息传递到全身:病原体的增殖,产生的多种酶、毒素及代谢产物等异种物质,通过对巨噬细胞和内皮细胞的激活,补体系统、激肽系统、凝血系统和纤溶系统的级联放大反应先后被触发。导致各种炎症介质如补体活化成分C5a、缓激肽、TNF-α、白细胞介素-6(IL-6)、前列腺素E 2 (PGE 2 )、NO、溶酶体酶等的生成,产生相应效应,主要包括血管扩张、血管通透性升高、细胞激活/趋化/黏附、凝血等。血管扩张和微血管的通透性增加,增加损伤局部组织的氧和营养物的供应,产生红、肿、热、痛等典型临床表现。炎症反应产生的细胞因子,包括TNF-α、IL-1和IL-8等,充当生理信使的角色,吸引多形核白细胞(PMNs)、单核巨噬细胞等吞噬细胞进入损伤部位,激活的白细胞和内皮细胞表面黏附分子和受体的表达量显著增加,使得白细胞同内皮细胞结合而附壁,内皮细胞收缩使血管内皮间隙增大,有利于吞噬细胞的移行,促使吞噬细胞进入感染区域以清除病原体。同时,黏附分子的表达增加使得微循环中的血细胞由“轴流”变成“边流”,出现血细胞附壁、集聚,继而触发凝血级联反应,形成局部微血栓,从而减少血液的丢失,限制损伤进一步扩散,在生理上起到隔离炎症区域的作用。在此过程中,中性粒细胞主要发挥吞噬作用,单核-巨噬细胞通过产生细胞因子协助吞噬过程。局部炎症的初衷是一种机体保护性的防御机制,是一种组织损伤—修复的过程,但若损伤产生的刺激过于剧烈,炎症刺激级联放大,超出机体自身可控范围,将引起进一步的机体损伤,而这种损伤,通常较原发损伤更为强烈,对机体打击更大。
2.机体损伤时的全身性炎症反应 机体遭受损伤应激时,会产生一系列的心血管和神经内分泌方面的全身性改变,如心率加快、呼吸加快、心排血量增加以及儿茶酚胺、糖皮质激素和胰高血糖素等物质释放增加,使机体产生适应性的效应,表现为:起初氧消耗的增加,机体通过代偿,尚能维持足够的氧输送;当氧耗进一步加大,出现失代偿情况,将会导致氧债加大、无氧代谢增强、乳酸等代谢产物集聚,引起血管扩张。同时,肥大细胞被激活后释放组胺与5-羟色胺,也会进一步引起血管扩张。如若无二次损伤打击的出现,机体的全身炎症反应高峰改变通常发生在损伤后的数天内,并可延续7~10天;患者通过自发的尿量增加,第三间隙液体减少,心率和体温逐渐恢复至正常,预示着病情好转,并且临床过程将相对简单。
局部的炎症反应是机体损伤的保护性反应,它通常被严格控制损伤的局部,如果失去了局部的控制或过度的激活,常导致过度的全身炎症反应,即为SIRS。病原体及其代谢产物逃脱局部防御进入循环系统后,导致血管内补体和凝血系统的激活;全身促炎细胞因子的连锁反应,刺激中性粒细胞释放溶酶体酶,通过呼吸暴发产生氧自由基,旨在杀灭或吞噬病原微生物、分解坏死组织,但同时也可引起血管内皮细胞的损伤,组织因子暴露,引起血小板聚集,进一步促进凝血系统的活化,联合局部小血管的收缩,最终导致微血栓形成、微循环障碍的发生。全身水平的炎症反应启动,导致血流动力学改变,毛细血管渗漏,全身水肿,进一步加重局部的微循环障碍,导致组织破坏。局部炎症可通过促炎介质使得循环中及远隔部位的中性粒细胞、巨噬细胞激活,如肝内的库普弗细胞等,导致远隔脏器的损伤,损伤的远隔脏器又将成为新的炎症反应策源地,呈现出级联放大的效应,最终引起瀑布式的炎症因子风暴,形成恶性循环。
Bone将SIRS的发展分为3个阶段:第一阶段,针对损伤局部的炎症反应,在局部环境中产生少量的细胞因子,有利于创伤修复和募集单核-吞噬细胞系统的细胞。第二阶段,少量细胞因子从损伤局部被释放到循环中,以增加局部的炎症反应:包括募集白细胞和血小板,刺激生长因子产生、启动急性相反应等,同时通过促炎介质和抗炎症介质之间的平衡,使炎症反应严格地被控制在适当的状态,直到损伤恢复。少部分患者进入第三阶段:此时,促炎介质和抗炎介质不能达到平衡,细胞因子的主要效应不是保护而是破坏作用,全身炎症反应开始启动,大量的炎症介质触发神经体液反应,同时丧失了微循环的完整性,并造成远处终末器官的损伤。
SIRS时,机体神经内分泌系统与免疫系统之间产生复杂的相互作用,炎症介质和效应细胞间的相互作用导致潜在的破坏性效应,归纳起来最主要包括以下几个方面:①白细胞和内皮细胞的激活;②实质细胞和免疫细胞的异常凋亡:内皮细胞凋亡;③外周血管扩张和血管通透性增加;④微血管内的凝血。
3.炎症反应的调控与失控 炎症是重要的防御反应,通常情况下,机体通过抗炎机制对炎症反应加以控制,避免过度的炎症反应对机体组织产生损害。炎症细胞的激活有明显的自限性,抗炎介质与促炎介质相互作用,发挥协调、抑制或相互拮抗的作用。当促炎反应占主导时表现为SIRS,当抗炎反应占主导时则表现为免疫抑制,即所谓的代偿性抗炎反应综合征(compensatory anti-inflammatory response syndrome,CARS),是1996年由Bone最先提出。CARS的发生主要与抗炎介质合成、抗炎内分泌激素释放及炎症细胞凋亡等因素有关。目前认为PGE 2 的持续释放是导致CARS的主要原因,糖皮质激素和儿茶酚胺也是参与CARS的重要因素。CARS的意义在于限制炎症,保护宿主免受过度炎症的损害。机体受细菌或毒素的刺激后,引起炎症细胞活化和炎症介质生成。与此同时,机体动员抗炎机制限制这种活化,这就是正常体内的炎症与抗炎症的平衡及其在机体自稳中的作用。当炎症刺激过强或持续刺激,则导致SIRS超过CARS,导致自身性破坏。反之,机体CARS过强,又可导致免疫功能低下,引起机会性感染的几率增大。维持“炎性免疫平衡”是临床治疗的目标。
CARS以机体免疫功能低下为特征,但难以进行临床判断。为使CARS应用于临床,1997年Bone提出CARS诊断标准:即外周血单核细胞HLA-DR表达量降至正常30%以下,且伴有炎症因子释放减少。初步的临床研究显示,符合SIRS的全身性感染患者,监测外周血单核细胞HLA-DR表达量,结果60%的患者符合CARS诊断标准。该结果难免使我们想到CARS的诊断标准是否与SIRS一样,灵敏度高而特异性低。与此同时,为了解释这一现象,Bone又将侵袭后的机体反应过程中的SIRS与CARS的并存状态称为混合性抵抗反应综合征(mixed antagonistic response syndrome,MARS),机体处于严重免疫紊乱的状况,临床上表现为持续广泛的炎症反应和免疫功能低下并存,导致MODS的发生和全身性的感染。
炎症反应的失控与促炎、抗炎效应的失衡有关,通常认为与炎症诱发因素的作用强度、持续时间以及炎症细胞的致敏有关。原发损伤使机体炎症细胞处于易被激活的致敏状态,轻度的“二次打击”即可再次引起的SIRS的发生。
4.参与SIRS的炎症介质及作用 炎症介质(inflammatory mediators)是在各种损伤因素的作用下,由病原体或受到刺激后的机体组织细胞、血浆产生和释放,参与或引起炎症反应的一类活性物质。狭义的炎症介质指的是“化学介质”,亦称之为“介质”。根据炎症介质的来源可分为:外源性的病原体及其代谢产物(内毒素等);细胞源性的介质包括生物活性胺、多种细胞因子、花生四烯酸代谢产物、PAF、NO等;血浆源性的介质包括补体系统、激肽系统、凝血系统、纤溶系统等。按照炎症介质的性质总的可分为促炎介质和抗炎介质,促炎介质主要包括:TNF-α、IL-1、IL-2、IL-6、IL-8、IL-12、NO、干扰素-γ(IFN-γ)、PAF、白三烯B4(LTB4)等,晚期促炎介质主要有高迁移族蛋白-1(HMGB-1);抗炎介质主要有IL-4、IL-5、IL-10、IL-13、PGE 2 、TGF-β等。炎症介质对血管内皮细胞和免疫炎性细胞的作用,在炎症过程中起着至关重要的作用;同时也因为它们对内皮细胞和免疫炎性细胞的过度活化,产生瀑布式的级联放大式的炎症反应,使炎症反应失去控制,导致SIRS的产生,因此,有人甚至认为SIRS是“介质病”。炎症介质的作用包括:①血管扩张,通透性升高,引起渗出;②对炎性细胞的趋化、激活作用;③导致组织损伤;④引起发热、疼痛等临床症状。
迄今为止对SIRS尚无理想的治疗方法。对SIRS治疗的目的和核心是及时有效地阻止SIRS向MODS转化。因此SIRS整体治疗的重点,应包括三个方面:原发病的治疗、器官功能的保护、预防“二次打击”。对可能出现的病情加重的因素进行干预,防止“二次打击”,对预防MODS的发生具有重要的意义。
1.原发病的治疗 尽管机体受损伤,SIRS被触发以后,沿着其自身的规律发展,其过程与原发损伤并无直接关系,但是,持续的损伤或再次的损伤无疑会加重SIRS,导致病情恶化。因此,妥善处理原发病,积极防治原发病的并发症,对SIRS的治疗具有根本的意义。
2.从整体的观念出发,维护器官功能 首先是液体复苏。机体遭受创伤感染或急性胰腺炎后,较早出现的全身病理变化往往就是低灌流和组织缺血缺氧。快速补充血容量,取得最佳前负荷,维持终末器官的灌注,从分子水平纠正缺氧状态,可以减轻缺血-再灌注损伤,是保护器官功能的重要措施。
当出现个别器官功能损害,内稳态不能维持时,应及时果断采取相应器官功能支持措施。低氧血症或呼吸困难的患者及时给予机械通气,有利于纠正组织缺氧,同时减轻患者呼吸运动的做功,减少全身的氧消耗,缓解全身氧供与氧耗的矛盾,对胃肠道黏膜的缺氧有保护作用,而胃肠道黏膜缺氧的纠正,可以减少内毒素的吸收,影响炎症反应过程,从而间接地减轻SIRS反应。同样,及时纠正血流动力学障碍,纠正酸碱、水、电解质的紊乱,维持患者适当的营养摄入,对其他器官功能的保护具有重要的意义。
3.预防“二次打击” 创伤感染烧伤等早期直接损伤作为第一次打击,所造成的全身炎症反应往往较轻,但第一次打击激活了机体的免疫系统,如果此后病情稳定,炎症反应往往就逐步减轻,患者康复。如果第一次损伤后再出现感染、休克、出血等第二次、第三次的打击,机体已处于激活状态的免疫系统,产生大量的炎症介质,导致组织器官更严重的损害,第二次打击强度本身可能不及第一次打击,但往往是致命性的。常见第二次打击包括感染、休克、出血、缺氧等,对治疗过程中可能出现的潜在发病因素施行预警性早期干预,防止“二次打击”,打断疾病恶性趋向化的链条,对预防SIRS转化为MODS具有重要的意义。
4.针对SIRS本身的治疗探索 二十多年来,针对SIRS有许多的治疗探索,抗介质治疗已被证实无效,血液滤过、免疫刺激等治疗近年似乎让人们看到希望,但至今并未被肯定,尚需进一步研究。
脓毒症的发生率高,是严重创伤、烧伤、休克和各种手术术后常见的并发症,全球每年约有2000万脓毒症病例,其中新生儿和儿童超过600万,每数秒钟即有一人死于脓毒症。在美国,每年约有75万例脓毒症患者,并且每年还以约9.0%的速度上升。脓毒症的病情凶险,病死率高,美国每年约21.5万人死亡,死亡率约28.7%。每年花费在脓毒症治疗上的费用高达167亿~200亿美元。据国外流行病学调查显示,脓毒症的病死率已经超过心肌梗死,成为重症监护病房内非心脏病患者死亡的主要原因。在我国,每年约有300万例脓毒症患者,对1991—2001年我国数家大型医院收住ICU的危重患者做回顾性调查发现,脓毒症死亡率高达28.6%~55.8%。近年来,尽管在抗生素治疗和器官功能支持手段等领域取得了长足的发展,但脓毒症的病死率仍然居高不下。脓毒症患者住院时间长,且需在ICU内接受长时间的强效治疗,医疗资源消耗巨大,已对人类健康造成了巨大威胁,并成为家庭和社会的严重负担。
脓毒症通常发生在严重创伤后的感染及各种化脓性感染,如烧伤、骨折、胆道及尿路感染等。病原微生物摆脱局部感染灶的限制进入血液循环中,或局部感染产生大量的炎症介质,引发SIRS的发生,形成脓毒症。此外,近年来肠道细菌易位的理念也逐渐为人们所认识,已经成为ICU患者发生脓毒症的重要病因。
脓毒症的风险因素主要包括:①原发病灶处理不当:脓肿引流不彻底,使得病原微生物的刺激持续存在。②机体抵抗力减弱:如存在慢性肾病、营养不良、老年患者等。③免疫功能低下:长期使用皮质激素、免疫抑制剂、新生儿等。④导管相关性感染:这已经成为急诊及ICU患者重要的感染源,管路表面为病原体的繁殖提供了很好的场所,导致“菌膜”的形成,进而激发SIRS,包括长期气管插管导致呼吸机相关性肺炎、中心静脉置管导致血源性感染、导尿管放置时间过长导致尿路感染等。⑤长时间高级别广谱抗生素的使用:在杀灭病原菌的同时也对机体内正常的共生菌造成损害,引发菌群失调,导致非致病菌或条件致病菌的大量繁殖,如艰难梭状芽孢杆菌( Clostridium Difficile )感染等。⑥长时间不经肠道喂养:肠上皮细胞约30%的营养由血液循环提供,70%依靠肠道上皮直接从肠腔内吸收,原发疾病引发的SIRS导致胃肠道血供减少,若肠道黏膜再长时间得不到营养物的供给,肠道黏膜将发生萎缩,肠黏膜屏障遭到破坏,导致细菌易位,肠源性感染。
导致脓毒症的病原微生物种类繁多,G - 菌中常见的有肠杆菌属、假单胞菌属等,G + 则为葡萄球菌、链球菌、肠球菌等,真菌主要为念珠菌感染多见。病原微生物的结构成分、代谢产物、内毒素或外毒素等均可导致SIRS的发生。
脓毒症具体的发病机制尚未明了,是机体受到病原微生物入侵后发生的一系列复杂的反应。虽然脓毒症是感染所引起,但一旦发生后,其发展遵循其自身的病理生理过程和规律,故从本质上讲,是宿主本身,而非病原体,介导了脓毒症的发病机制。脓毒症的临床表现和患者预后主要取决于初始的感染部位、引发感染的病原体、患者的基础健康状况、开始治疗的时机、器官功能障碍的方式等。其发生发展是受多基因调控的,涉及复杂的全身炎症反应、免疫功能障碍、凝血功能异常、组织损伤等多个方面,与机体多系统、多器官病理生理改变密切相关,脓毒症的发病机制仍需进一步阐明。
1.全身性过度炎症反应 脓毒症时,病原微生物侵入机体,激活机体单核巨噬细胞系统及其他炎症反应细胞,产生并释放大量炎性介质,引发SIRS。在抵御外界损伤因素入侵的同时,也造成机体各系统及器官的广泛损伤。
2.免疫抑制 脓毒症时CARS的发生,导致免疫功能低下,主要为丧失迟发性变态反应、不能清除病原体、继发感染的风险加大。脓毒症免疫功能紊乱的机制主要与CARS发生时抗炎介质的增多与免疫细胞异常凋亡有关。
3.肠道细菌和内毒素易位 20世纪80年代以来,随着人们对肠道功能认识的逐步加深,肠道是“多器官功能障碍的发动机”为人们所认可,其主要原因就是肠道细菌及内毒素易位所致。其主要机制表现在三个方面:SIRS时机体处于应激状态,肠道血供减少,肠黏膜屏障受损;长时间肠道喂养的缺失,进一步加重肠道黏膜上皮的萎缩,屏障功能进一步受损;脓毒症时长疗程高级别大剂量抗生素的使用,导致肠道菌群紊乱的发生。这些导致肠道微环境的严重破坏,引发细菌及内毒素的易位,引起继发感染的发生,二次打击导致进一步的全身性损害,导致器官功能障碍和机体损伤。
4.凝血功能紊乱 凝血系统在脓毒症的发病过程中起着重要作用,它与炎症反应相互促进,共同构成脓毒症发生、发展中的关键因素。而脓毒症又会进一步加重机体的凝血功能障碍,内毒素和TNF通过诱发巨噬细胞和内皮细胞释放组织因子,可激活外源性凝血途径,被内毒素激活的凝血因子Ⅻ也可进一步激活内源性凝血途径,最终导致DIC。
5.基因多态性 临床上常见受到同一致病菌感染的不同个体的临床表现和预后截然不同,有些表现为严重脓毒症甚至脓毒性休克,但有些患者无明显临床症状,只是单纯的菌血症。这些提示基因多态性等遗传因素也是影响人体对应激打击易感性与耐受性及药物治疗反应差异性的重要因素。
针对严重脓毒症和脓毒性休克对人类的巨大危害和挑战,2001年欧洲危重症学会(ESICM)、美国危重症学会(SCCM)和国际脓毒症论坛(ISF)发起“拯救脓毒症战役”(surviving sepsis campaign,SSC),2002年欧美国家多个组织共同发起并签署“巴塞罗那宣言”,计划在5年内将脓毒症患者的死亡率减少25%,并且制定基于对脓毒症研究的循证医学证据,并不断更新脓毒症治疗方案即拯救脓毒症指南。为改进脓毒症的治疗措施,降低脓毒症的死亡率。拯救脓毒症指南于2004年第一次制定,后分别于2008年、2012年进行了两次修订。并从2012年起将每年的9月13日定为“世界脓毒症日”,并建立了拯救脓毒症的官方网站:www.survivingsepsis.org/Pages/default.aspx。
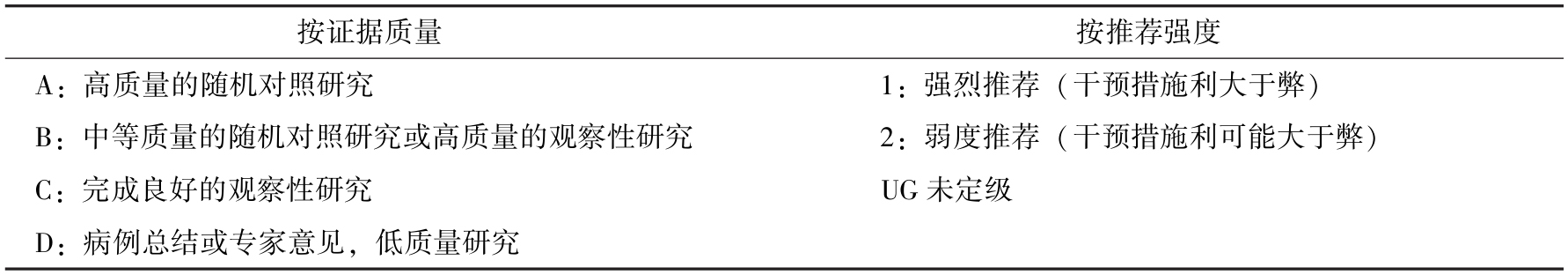
现就2012版拯救脓毒症指南GRADE(Grading of Recommendations Assessment,Development and E-valuation)中最新有关脓毒症及脓毒性休克管理的推荐汇总于表15-4:
表15-4 GRADE指南推荐等级
A.早期复苏
1.初始复苏:仍然依据早期目标导向性治疗(early goal-directed treatment,EGDT)的标准,对于感染性休克,应尽早识别组织低灌注的患者并尽快转入ICU,在复苏的第1个6小时,复苏目标:CVP 8~12mmHg,MAP≥65mmHg,尿量≥0.5ml/(kg·h),ScvO 2 ≥70%或SvO 2 ≥65%(1C)。
2.如果以血乳酸升高作为组织低灌注的指标,应尽快复苏使血乳酸回归正常(2C)。
B.脓毒症及其改善的筛查
1.对可能存在感染的重症患者常规筛查重度脓毒症以便及时诊断和早期治疗(1C)。
2.将改善重度脓毒症管理的成果应用于改善患者预后(UG)。
C.诊断
1.应用抗生素前,采集适当的细菌学标本,并尽可能在45分钟内完成(1C);血培养至少为双份(需氧和厌氧培养皿),应分别经皮穿刺和经血管内导管(除非导管留置时间<48小时)抽取标本,其他部位感染时也应在给予抗生素前留取标本,如尿、脑脊液、伤口或其他体液(1C)。
2.需要对侵袭性念珠菌病进行鉴别诊断时可检测1,3β-D葡聚糖(2B)甘露聚糖和抗甘露聚糖抗体(2C)。
3.及早进行影像学检查以明确感染灶(UG)。
D.抗生素治疗
1.在发现脓毒性休克(1B)和无休克的重度脓毒症(1C)后,应将1小时内静脉应用有效抗生素作为治疗的目标。
2a.初始经验性抗感染治疗应包括一种或多种抗生素:能够覆盖所有可能的病原体(细菌、真菌或病毒),且能够在诱发脓毒症的感染部位达到充分的药物浓度(1B)。
2b.应每天评估抗生素方案以寻求降阶梯治疗的可能(1B)。
3.低水平的降钙素原或类似生物标记物可帮助临床医师对有脓毒症但无其他感染证据的患者停止经验性抗生素治疗(2C)。
4a.根据个体患者的现存疾病和当地感染类型选择能覆盖最可能病原体的抗生素进行经验性治疗。对中性粒细胞减少的严重感染患者(2B)、难治性的多重耐药菌如不动杆菌属和假单胞菌属感染的患者(2B)建议经验性抗生素联合治疗;严重感染伴呼吸衰竭和脓毒性休克的患者,建议超广谱β内酰胺类抗生素联合一种氨基糖苷类或氟喹诺酮类抗生素治疗铜绿假单胞菌血症(2B);肺炎链球菌感染导致的脓毒性休克可应用β内酰胺类联合大环内酯类抗生素(2B)。
4b.经验性抗生素联合治疗重症感染建议不要超过3~5天,当得到药敏结果后,尽快选择最合适的单药进行降阶梯治疗(2B)。
5.一般建议抗生素治疗时间为7~10天,对治疗反应缓慢、感染病灶无法通畅引流、金黄色葡萄球菌血症、某些真菌和病毒感染、免疫缺陷包括中性粒细胞减少的患者可延长疗程以获得充分治疗(2C)。
6.病毒导致的严重脓毒症、脓毒性休克患者尽早开始抗病毒治疗(2C)。
7.对非感染原因导致的严重炎症状态不推荐应用抗生素(UG)。
E.感染源控制
1.为及时控制感染源需要尽快寻找并明确解剖学诊断或排除诊断,并在诊断后12小时内进行干预(1C)。
2.当胰周坏死组织可能为感染源时,建议等待坏死组织和正常组织分界明确后再进行确定性的干预(2B)。
3.当感染源需要处理时,最好采用对机体干扰最小的有效干预措施,如经皮穿刺引流而不选择手术切开引流脓肿(UG)。
4.当血管内导管可能是严重脓毒症或脓毒性休克的感染源时,建立新的血管通路后立即拔除导管(UG)。
F.感染预防
1.建议应用和调研选择性口咽去污(selective oral decontamination,SOD)和选择性消化道去污(selective digestive decontamination,SDD)作为减少VAP发生的方法。这些感染控制的措施可用于方法学上已被证实有效的健康护理单位和病房(2B)。
2.葡萄糖酸氯己定可用于口咽去污以降低ICU严重脓毒症患者VAP的发生率(2B)。
G.严重脓毒症的液体治疗
1.首选晶体液进行液体复苏(1B)。
2.不推荐羟乙基淀粉(HES)用于重度脓毒症和脓毒性休克的液体复苏(1B)。
3.重度脓毒症和脓毒性休克需大量晶体液时可加用白蛋白进行液体复苏(2C)。
4.脓毒症导致的组织低灌注并怀疑低血容量时,初始液体负荷试验至少给予30ml/kg晶体液(可部分为白蛋白等效液),部分患者可能需要快速大量补液(1C)。
5.只要补液试验后动态(如:脉压改变、每搏量)或静态(如:动脉压及心率的变化)参数显示血流动力学有改善,就应该继续进行补液(UG)。
H.血管活性药物
1.升压药治疗起始目标为MAP≥65mmHg(1C)。
2.首选去甲肾上腺素(1B)。
3.需要额外的药物维持适当血压时,建议给予肾上腺素(联合或替代去甲肾上腺素)(2B)。
4.可使用血管加压素(vasopressin,0.03U/min)以升高MAP或减少去甲肾上腺素用量(UG)。
5.不推荐单纯应用小剂量血管加压素初始治疗脓毒症低血压,高于0.03~0.04U/min剂量的加压素用于补救治疗(其他升压药不能维持适当的MAP)(UG)。
6.多巴胺仅限于快速心律失常风险低和绝对或相对心动过缓的患者(2C)。
7.不推荐苯肾上腺素应用于脓毒性休克的治疗,下述情况除外:去甲肾上腺素引起严重心律失常;心排血量高伴顽固性低血压;作为强心药/升压药物联合小剂量血管加压素仍不能维持目标MAP的补救治疗(1C)。
8.不推荐小剂量多巴胺用作对肾的保护治疗(1A)。
9.如果条件允许,推荐所有需要升压药的患者进行动脉置管,监测血流动力学(UG)。
I.正性肌力药物
在心脏充盈压升高而心排血量低提示心肌功能障碍时或经充分补液并达适当的MAP时仍存在组织灌注不足的征象,推荐尝试输注多巴酚丁胺20μg/(kg·min),必要时与升压药联合应用(1C)。
J.皮质类固醇激素
1.如果脓毒性休克成年患者经充分液体复苏和升压药治疗能够恢复稳定的血流动力学,则不建议应用静脉氢化可的松;若不能达到血流动力学目标时建议每天给予静脉氢化可的松200mg(2C)。
2.不建议使用ACTH激发试验来判断成年感染性休克患者是否需要氢化可的松(2B)。
3.当不再需要升压药时建议逐渐停用激素(2D)。
4.脓毒症患者在没有出现休克时不推荐使用皮质类固醇激素(1D)。
5.建议持续输注小剂量氢化可的松,而不分次推注(2D)。
K.血制品的应用
1.纠正组织低灌注后,且不存在削弱组织氧合的情况,如心肌缺血、严重低氧血症、急性出血或缺血性心脏病等,仅当血红蛋白低于70g/L时再输注红细胞,使成人患者血红蛋白达70~90g/L(1B)。
2.不推荐应用红细胞生成素作为严重脓毒症贫血的治疗策略(1B)。
3.在没有出血或计划行侵入性操作时,建议新鲜冰冻血浆不用于纠正实验室凝血指标紊乱(2D)。
4.不使用抗凝血酶治疗严重脓毒症和脓毒性休克(1B)。
5.严重脓毒症患者血小板<10×10 9 /L时,即使无明显出血,也需预防性输注血小板;当血小板<20×10 9 /L且有明显出血风险时可考虑输注血小板;在活动性出血手术或有创操作之前要求血小板达到较高水平(50×10 9 /L)(2D)。
L.免疫球蛋白
严重脓毒症或脓毒性休克成年患者不建议静脉应用免疫球蛋白(2B)。
M.硒
严重脓毒症患者不建议静脉应用硒治疗(2C)。
N.活化蛋白C的推荐已成为历史
活化蛋白C已退市。
2004年和2008年指南对重度脓毒症应用活化蛋白C均有推荐,随后的临床研究不支持活化蛋白C用于重度脓毒症患者,药品已撤出市场。
O.脓毒症导致的ARDS的机械通气策略
1.推荐脓毒症导致的ARDS患者的目标潮气量是6ml/kg(1A.vs.12ml/kg)。
2.推荐监测ARDS患者的平台压,被动通气时平台压≤30cmH 2 O(1B)。
3.推荐使用PEEP避免呼气末肺泡萎陷(1B)。
4.脓毒症导致的中、重度ARDS患者建议应用较高水平的PEEP(2C)。
5.对于顽固性低氧血症患者,建议进行肺复张(2C)。
6.氧合指数≤100mmHg的ARDS患者,条件允许时可进行俯卧位通气(2B)。
7.推荐机械通气的脓毒症患者床头抬高30°~45°以降低误吸风险,预防VAP发生(1B)。
8.少数脓毒症诱发ARDS患者,经认真评价无创通气的优点且认为利大于弊的情况下,可应用无创面罩机械通气(2B)。
9.推荐对机械通气的重度脓毒症患者制定脱机预案,有规律地进行自主呼吸试验评估撤机的可能性;安全撤机标准为:可唤醒;血流动力学稳定(未应用升压药);无新发潜在危险因素;较低的通气支持压力及PEEP;较低的吸氧浓度,可更换为面罩或鼻导管吸氧。如果自主呼吸实验成功,可考虑拔管(1A)。
10.脓毒症导致的ARDS患者反对常规使用肺动脉导管(1A)。
11.脓毒症诱发ARDS的患者无组织灌注不足时推荐限制性补液,尽可能的“干”(1C)。
12.无特殊指征如支气管痉挛时不推荐应用β 2 受体激动药治疗脓毒症诱导的ARDS患者(1B)。
P.脓毒症时的镇静 、 镇痛和肌松
1.机械通气脓毒症患者推荐最低剂量进行持续或间断镇静,并逐渐调整剂量至镇静目标(1B)。
2.无ARDS的脓毒症患者应尽量避免使用肌松药,如果患者必须使用肌松药,可按需间断静脉推注或在4个成串刺激监测阻滞深度下持续输注(1C)。
3.脓毒症导致的氧合指数<150mmHg的早期ARDS患者可短程(≤48小时)应用神经肌肉阻滞药(2C)。
Q.血糖控制
1.对ICU脓毒症患者制定血糖管理预案,当连续2次血糖>10mmol/L时开始应用胰岛素,血糖目标水平≤18mmol/L,而非6.1mmol/L,即不推荐强化胰岛素治疗(1A)。
2.接受胰岛素控制血糖的患者需每1~2小时监测血糖,血糖水平和胰岛素输注速度相对稳定后可每4小时监测血糖(1C)。
3.谨慎解读床旁检测的毛细血管血糖结果,此法不能精确估计动脉血或血浆葡萄糖水平(UG)。
R.肾替代治疗
1.建议持续肾替代治疗或间断血液透析均可用于严重脓毒症和急性肾衰竭患者,两种方法的患者短期生存率相似(2B)。
2.血流动力学不稳定的脓毒症患者建议使用持续肾替代治疗以便液体平衡管理(2D)。
S.碳酸氢钠治疗
对灌注不足导致的pH≥7.15的乳酸酸血症,不推荐以改善血流动力学和减少升压药需求为目的而应用碳酸氢钠(2B)。
T.深静脉血栓的预防
1.推荐重度脓毒症患者每天应用药物预防静脉血栓栓塞(venous thromboembolism,VTE)(1B);可每天1次皮下注射低分子肝素(low molecular weight heparin,LMWH)(1B:与每天2次肝素相比较;2C:与每天3次肝素比较)。如果肌酐清除率<30ml/min,推荐应用肝素(1A)或其他经肾代谢程度低的LMWH(2C)或者普通肝素(1A)。
2.建议重度脓毒症患者采取药物和间歇充气加压袋联合预防(2C)。
3.有肝素禁忌证(如血小板减少、严重凝血病、活动性出血、近期颅内出血等)的脓毒症患者不推荐药物预防(1B);如无相关禁忌证则建议应用机械性预防措施,如分级加压弹性长袜或间歇性加压装置(2C);风险降低后建议开始药物预防(2C)。
U.应激性溃疡的预防
1.存在出血危险因素的严重脓毒症/脓毒性休克患者推荐使用质子泵抑制剂(PPI)或H 2 受体阻滞药(H 2 RA)预防应激性溃疡(1B)。
2.预防应激性溃疡首选PPI而非H 2 RA(2D)。
3.无出血危险因素的患者可以不采取预防用药(2B)。
V.营养
1.诊断严重脓毒症或脓毒性休克后48小时内,如果可耐受,可行口服或肠内喂养,而不是完全禁食或仅静脉应用葡萄糖(2C)。
2.第1周内避免强制性供给全热量,而应采取低热量喂养[如每天2092kJ(500kcal)],耐受后逐渐加量,即“允许性低热量”(2B)。
3.诊断严重脓毒症或脓毒性休克7天内可应用静脉内葡萄糖和肠内营养,而不是单纯的全肠外营养或肠外肠内联合喂养(2B)。
4.严重脓毒症患者营养支持无须补充特殊免疫调节物质(2C)。
W.治疗目标的确立
1.与患者和家属沟通治疗和预后目标(1B)。
2.管理目标与治疗和临终关怀计划相结合,必要时采用姑息治疗原则(1B)。
3.入ICU 72小时内尽早与家属沟通治疗相关事宜(2C)。
脓毒症集束化(bundle)管理情况见表15-5。
表15-5 脓毒症集束化(bundle)管理

∗滴定式复苏目标包括:CVP≥8mmHg、ScvO 2 ≥70%及乳酸正常
( 李维勤 邹 磊 )
Artigas A, Niederman M S, Torres A, Carlet J. 2012. What is next in sepsis: current trials in sepsis[J]. Expert Rev Anti Infect Ther, 10 (8): 859-862.
Bone R C. 1996. Sir Isaac Newton, sepsis, SIRS, and CARS[J]. Crit Care Med, 24 (7): 1125-1128.
Dellinger R P, Levy M M, Rhodes A, et al. 2013. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012[J]. Crit Care Med, 41 (2): 580-637.
Derek C, Angus M D, Tom van der Poll. 2013. Severe Sepsis and Septic Shock[J]. N Engl J Med, 369: 840-851.
Fink M P, Evans T W. 2002. Mechanisms of organ dysfunction in critical illness: report from a Round Table Conference held in Brussele[J]. Intensive Care Med, 28: 369-375.
Hotchkiss R S, Karl J E. 2003. The Pathophysiology and Treat-ment of Sepsis[J]. N Engl J Med, 348: 138-150.
Levy M M, Fink M P, Marshall J C, et al. 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference[J]. Intensive Care Med, 29: 530-538.
Members of the American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference Committee. 1992. Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis[J]. Crit Care Med 20: 864-874.
Moyer M W. 2012. New biomarkers sought for improving sepsis management and care[J]. Nat Med, 18 (7): 999.
Vincent J L, Opal S M, Marshall J C, et al. 2013. Sepsis definitions: time for change[J]. Lancet, 381 (9868): 774-775.