
下载掌阅APP,畅读海量书库
立即打开



分子遗传控制个体对疾病的易感性,而表观遗传则最终决定疾病的发生和表型,因而表观遗传学在酒精依赖的病理生理学中起重要作用。酒精可通过表观遗传学机制干扰个体体内一些化学过程。研究提示表观遗传学机制,如组蛋白乙酰化、磷酸化、甲基化及DNA甲基化,参与包括成瘾行为在内的脑损害的病理生理学。除此之外,研究者还针对胎儿酒精综合征和酒精性肝病等表观遗传学机制进行了研究。
大量饮酒可能导致人类精液中DNA超甲基化区域甲基化程度降低。1991年,研究者针对酒精及其代谢产物乙醛对幼鼠DNA甲基化的影响进行了研究,发现孕鼠被灌酒精后,幼鼠DNA呈现低甲基化状态,甲基化转移酶的活性也降低,体外试验也发现乙醛可抑制DNA甲基化转移酶的活性,见图2-2-1。虽然研究提示酒精依赖患者的整体(基因组)DNA超甲基化,但并不是所有的基因都呈现超甲基化,如早老素1基因。研究显示酒精依赖患者某些候选基因启动子特异性DNA甲基化发生改变,具体如下:
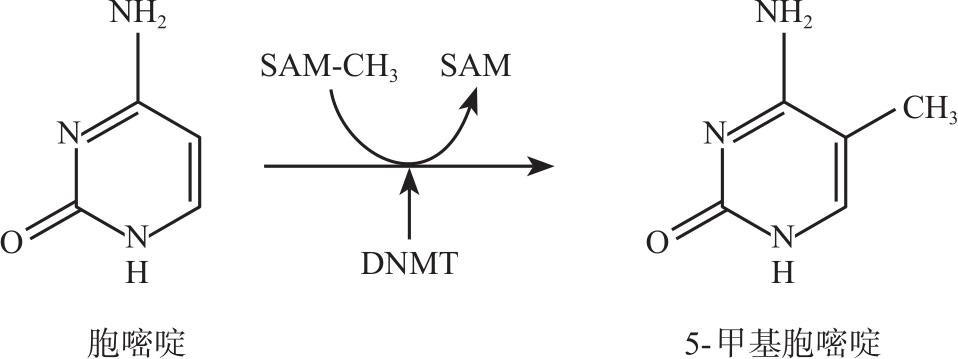
图2-2-1 DNA甲基化修饰
α突触核蛋白在多巴胺神经递质传递中起重要作用,可调节突触间多巴胺平衡,影响多巴胺合成基因的表达,因此多巴胺神经递质循环是调节酒精依赖戒断和渴求的重要机制。α突触核蛋白启动子区DNA甲基化增高,α突触核蛋白低表达,导致多巴胺神经递质传递紊乱,提示表观遗传修饰也许能解释为什么酒精依赖患者在饮酒状态下会对酒精的渴求度下降。但有研究却显示酒精依赖大鼠模型不同脑区α突触核蛋白表达增高,酒精依赖患者α突触核蛋白mRNA表达也同样增加。
表观遗传学研究揭示的另一个机制是酒精依赖与内质网应激的连接。HERP是一种内质网固有膜蛋白质,主要存在于发育的神经元和成人脑组织中,可调节Ca 2+ 平衡,保护内皮和神经元细胞完整性,对抗氧化性应激,在内质网应激状况下起神经保护作用。酒精依赖患者同型半胱氨酸(homocysteine,HCY)升高,导致内质网应激,与此同时血中HERP启动子区甲基化增高,HERP mRNA表达降低,HERP低表达可能导致动脉事件、痉挛发作和其他神经学损伤。
加压素和ANP等激素水平变化可影响酒精依赖患者对酒精的渴求度,这种影响受表观遗传学修饰。加压素有致焦虑的作用,与饮食调节和强迫行为有关;ANP有抗焦虑作用,与酒精戒断有关系。酒精依赖患者白细胞(white blood cell,WBC)中加压素和 ANP 基因表达与表观遗传修饰有关,研究显示与健康者相比,酒精依赖患者加压素基因启动子区超甲基化,但其mRNA表达未见统计学意义,而ANP启动子区甲基化程度降低,其mRNA表达增高。
多巴胺神经递质在酒精依赖产生和维持中起重要作用。DAT促进突触间隙多巴胺的重摄取,增加患者对酒精的渴求度。研究提示,酒精依赖患者多巴胺受体的活性降低,但戒酒后其又恢复正常。酒精依赖患者DAT启动子区特异性甲基化可能影响多巴胺神经传递,与健康者相比,酒精依赖患者DAT启动子区超甲基化,且在戒酒过程中DAT启动子也发生表观遗传改变,提示DAT启动子超甲基化可能在多巴胺神经传递中起重要作用,与酒精依赖患者渴求度增加有关。以上研究提示酒精依赖患者DAT启动子区超甲基化,降低了DAT mRNA的表达水平,致使突触间隙多巴胺聚集,进而降低了酒精依赖患者对酒精的渴求。
最近的研究显示表观遗传学修饰是小鼠皮质神经元 NR2B 基因转录的重要调控机制。酒精可增加成人皮质和胎儿皮层神经元中 NR2B 基因表达,提示NR2B表达受DNA低甲基化调控。慢性酒精暴露可引起 NR2B 基因内含子1和内含子2区呈现低甲基化,NR2B亚型表达上调与 NR2B 基因CpG岛甲基化降低有关,然而急性酒精中毒时,既不影响DNA甲基化也不改变 NR2B 基因表达。虽然慢性酒精中毒可上调 NR2B 基因表达,但是酒精戒断48h后 NR2B 水平又恢复正常。
最近一项关于145例酒精依赖患者和37名健康者 POMC 基因5′端启动子区甲基化的研究显示, POMC 基因启动子甲基化与酒精依赖患者的渴求度有关。因为 POMC 基因编码一种激素前体,在下丘脑-垂体-肾上腺(the hypothalamic-pituitary-adrenal axis,HPA)轴中起重要作用,提示酒精摄入可引起表观遗传改变,可能导致HPA轴功能失调引起患者对酒精渴求。
外周血中HCY浓度增高可影响基因组DNA的甲基化状态。研究表明酒精依赖患者基因组DNA甲基化增高(10%),与HCY浓度增高关系密切。非戒断期酒精依赖患者血浆HCY浓度与血液中酒精浓度密切关系,而戒酒期间HCY水平会持续下降。
另外,一些研究者证实表观遗传学机制,特别是DNA甲基化模式的改变可调控胆碱对学习和记忆的作用。一项对191例美国艾奥瓦州寄养子受试者 MAOA 启动子区甲基化状态的研究发现, MAOA 甲基化状态与女性酒精依赖、烟草滥用有关,而与男性无关。另外一项针对韩国男性酒精依赖患者与健康者 5-HTT 启动子区甲基化状态的研究显示,两组甲基化水平无差异。与健康组相比,酒精依赖患者DNA甲基化基因( DNMT )—— DNMT3a 和 DNMT3b mRNA表达降低。这一降低与血中酒精浓度和DNA甲基化增高呈正比,提示 DNMT mRNA表达降低导致DNA甲基化,进而影响基因表达。不同基因甲基化状态的改变可解释酒精诱发疾病或症状的发病机制。
酒精不仅影响DNA甲基化,而且也可影响组蛋白修饰。一项关于肝脏星形细胞中酒精对组蛋白乙酰化作用的研究提示,酒精可使 H3L9 、 H3L23 乙酰化增加。一项研究提示酒精可引起组蛋白某些位点选择性乙酰化、甲基化和磷酸化。
酒精可增加组蛋白乙酰转移酶(HATs)的活性。酒精可引起小鼠肝脏原代培养中 H3K9 乙酰化,这种现象在肝脏星形细胞和用大量酒精处理后的小鼠组织中(例如肺、脾、睾丸等)也被观察到。大鼠酒精灌胃1个月后,其肝脏中有多于1300种基因表达发生改变。酒精导致肝脏缺氧,增加了核苷酸信号转录因子HIF-1α-β,反过来增强了P300的活性,也增强了H3K9乙酰化和基因转录活性。乙醛和乙酸盐是酒精的代谢产物,能引起H3K9乙酰化。但是在这个阶段,酒精或乙酸盐诱导组蛋白乙酰化可能是由不同途径导致的,因为酒精的作用可被促分裂原活化蛋白(mitogenactivated protein kinase,MAP)激酶抑制剂抑制。如果说酒精和/或乙醛增强了组蛋白乙酰化,导致 ADH1 基因表达增强,反过来, ADH1 基因表达增强也将增加酒精的代谢产物。
研究提示摄入酒精所致的抗焦虑作用以及戒断引起的焦虑症状都可由表观遗传现象触发。在这个机制中,与环腺苷酸反应结合蛋白(cAMP response element-binding protein,CREB)有关的神经肽Y(neuropeptide Y,NPY),可作为内源性抗焦虑成分。大量摄入酒精时,产生抗焦虑作用,原因是组蛋白去乙酰化酶(histone deacetylase,HDAC)抑制剂致使H3和H4乙酰化增加,CREB水平增加,大鼠杏仁核区NPY表达增高。另一方面,戒断引起的焦虑与组蛋白H3和H4乙酰化降低有关,组蛋白H3和H4乙酰化降低是由HDAC活性增加、CREB水平和NPY表达降低引起的。用HDAC抑制剂、制滴菌素A(trichostatin A,TAS)治疗大鼠戒酒症状,增加了NPY的表达,大鼠的焦虑样行为减少。以上研究不仅为酒精依赖和戒断症状提供了一个可能机制,还提示HDAC抑制剂可能是治疗酒精依赖的新方向。
研究发现大鼠肝脏原代培养中,酒精可降低H3K9甲基化和基因表达,例如Lsdh和细胞色素P4502c11,同时增加H3K4甲基化,上调 ADH 和 GST-yc2 等基因表达,提示表观遗传修饰可改变基因表达,继续饮酒可导致脂肪肝、炎症和肝硬化等发生。另一项关于肝脏内质网应激通路的表观遗传调控酒精性脂肪肝发病机制的研究报告显示,酒精降低 GRP78 、 SREBP-1C 、 GADD153 等基因启动子区的H3K9甲基化水平,进而上调内质网应激信号。
组蛋白乙酰化与甲基化错综复杂,有时调控作用甚至相反。肝细胞H3K4甲基化与基因表达上调有关,H3K9甲基化与基因表达下调有关。一方面,酒精可引起H3K9乙酰化增高,降低H3K9甲基化,同时也可引起H3K4甲基化增高。实验证实酒精引起的H3K4甲基化与基因表达(谷胱甘肽-S-转移酶-yc2)上调有关,而H3K9甲基化与基因表达(L-丝氨酸脱水酶,cytP4502c11)下调有关。因此,组蛋白H3不同赖氨酸残基甲基化导致的转录反应可能不同。
组蛋白磷酸化在酒精依赖中也被涉及。研究者发现酒精诱导的肝毒性是通过有丝分裂原活化蛋白激酶信号通路调节发生的。组蛋白H3磷酸化作为这种调节机制可能带来后果的相关研究显示,大鼠肝细胞原代培养中,酒精及其代谢产物乙醛增加了H3丝氨酸的磷酸化,p38促分裂原活化蛋白激酶磷酸化和蛋白水平增高,导致H3磷酸化增高。
除了上述定义的表观遗传学机制之外,微RNA(miRNA)通过对基因表达转录后调节来传达表观遗传样特征。miRNA通过靶向降解mRNA或特异性抑制mRNA转录来快速调节基因表达。研究证实酒精可调控钙离子激活的K + 通道(BK通道)表达,导致患者酒精耐受性增加。最近关于酒精耐受性的研究涉及mRNA在酒精诱导BK通路功能改变中的作用。一项研究提示急性酒精中毒会使BK通路表达急剧下调,同时miRNA-9表达上调。有趣的是,miRNA-9转录后调控BK mRNA剪接体编码BK通道亚型,使其显示不同的酒精依赖易感性。这些研究提示miRNA在内的转录后表观遗传学修饰可引起酒精耐受增加。另外,研究发现酒精可抑制四种miRNA,即miRNA-21、miRNA-335、miRNA-9、miRNA-153,它们可能与酒精所致胎儿畸形有关。
近年来研究者针对胎儿酒精综合征(fetal alcohol syndrome,FAS)的表观遗传学机制进行了研究,表明胎儿酒精谱系障碍(FASD)潜在的表观遗传学机制包括酒精诱导的甲基代谢修饰,导致发育过程中DNA甲基化模式和基因表达发生改变。早期神经胚形成过程中,酒精暴露诱导的DNA甲基化变化与基因表达改变、FASD发病有关。研究显示酒精可阻止重要的神经干细胞的DNA甲基化编程,阻止神经干细胞分化。在FASD的动物模型中,胚胎植入阶段酒精暴露可改变胎盘特异区域的甲基化状态,但不能改变胚胎的甲基化状态。
表观遗传机制在酒精性肝病(alcoholic liver disease,ALD)的进展中起重要作用。过去几十年,一些研究证实酒精性肝病肝脏中的表观遗传修饰发生改变,包括DNA甲基化、组蛋白甲基化、miRNA及甲基化转移酶(DNMTs)、HATs、组蛋白去乙酰化酶(HDACs)。腺苷蛋氨酸(SAMe)是体内重要的甲基供体。酒精性肝病患者肝脏中SAMe耗竭,导致高同型半胱氨酸血症,影响DNA甲基化。用酒精灌胃9周后,大鼠肝脏中甲硫氨酸、SAMe、谷胱甘肽含量降低,DNA甲基化也减少40%;酒精除了对DNA低甲基化有直接影响外,其代谢产物乙醛对DNMTs和甲硫氨酸合成酶也有直接影响;与基因转录和/或沉默有关的疾病发展过程中,DNA甲基化与组蛋白乙酰化相互作用。例如靶基因启动子区CpG岛超甲基化导致局部染色体去乙酰化,反之,组蛋白乙酰化水平降低可能对DNA甲基化敏感。慢性酒精中毒的肝脏中H3K9超乙酰化、H3K9低甲基化和H3K4超甲基化相互存在;miRNA在类似酒精性脂肪肝等肝脏疾病中起重要作用。研究提示酒精性肝病中,酒精暴露可调控miRNA,进而控制转录后调控,影响基因表达。慢性酒精暴露可改变miRNA,进而影响肠的通透性。随着表观遗传学及其研究技术的飞速发展,人们对酒精依赖的发病机制有了更深刻的认识,为酒精依赖的诊断、预防和治疗提供了新的方向。
(施梅)