
下载掌阅APP,畅读海量书库
立即打开



卵巢是女性的主要生殖内分泌器官,不仅为人类繁衍提供配子,也能分泌多种激素参与全身多系统功能的支持和维护。它的一生经历着发生、成长、成熟和衰退及衰竭的不同阶段,体现为女性生殖能力的改变和激素水平的变化。女性卵巢一生的生理变化,与卵巢中的卵泡数量和质量息息相关,从胚胎期原始生殖细胞迁移至生殖嵴形成始基卵泡池开始,虽然最高峰时卵巢中含有600万~700万个始基卵泡,但与此同时始基卵泡因自发性的激活和闭锁而不断减少,至出生时仅剩100万~400万个始基卵泡。出生后始基卵泡仍然持续被募集,在出生后4周内受胎盘及母体性腺产生的雌激素影响,女性卵巢中的部分初级卵泡可继续发育,但此时的卵巢几乎无生理功能。此后卵巢进入缓慢的发育成长过程,从出生后4周到青春前期,绝大多数卵泡(平均50%~70%)在各个阶段发生退化而闭锁。直至青春期,卵巢在下丘脑和垂体的协助下逐渐发育成熟,开始为女性提供成熟的卵子并分泌激素和细胞因子,发挥成熟卵巢的生理功能,维持女性生育及多脏器功能。但随着卵泡数量持续减少和质量不断下降,围绝经期卵巢功能开始加速下滑直至卵泡池最终耗竭,功能丧失,从而引起绝经期综合征及全身多脏器功能损害,严重危害女性健康。
卵巢组织的发生可分为性未分化期和性分化期。两性别性腺在胚胎发育阶段初期并不具备明显区分的特征,但随后雌性胚胎在发育过程中,受到一系列关键基因及转录因子的作用,逐步分化为卵巢。
人类胚胎的遗传性别是由卵母细胞(oocyte)受精的精子种类(X或Y)所决定的。直到人胚胎第7周,生殖腺才开始有性别的形态学特征,男性和女性性腺的组织学特征才开始显现出来。早期两性别的生殖系统是相似的,在人类的早期胚胎均有向两性别分化的潜能。因此,生殖系统发育的早期阶段是性别发育的未分化的阶段。
原始性腺的发生始于生殖嵴的形成。生殖嵴为一对纵向嵴,来源于中胚层的中间带和其上覆盖的上皮,起初不含任何生殖细胞。在人胚胎第4周时,生殖细胞开始通过后肠的背系膜从卵黄囊的内胚层迁移到生殖嵴。在人胚胎第6周时达到生殖嵴。同时,生殖嵴的上皮增殖并穿透中间中胚层以形成原始性索,生殖细胞和原始性腺的组合形成了未分化的性腺,从而发育成睾丸或者卵巢。
约在人胚发育第4周,中胚层逐渐向腹侧移动,并与体节分离,形成左右两条纵行的索状结构,称为生肾索(nephrogenic cord)。在人胚发育第4周末,生肾索体积不断增大,从胚体后壁突向体腔,在背主动脉两侧形成左右对称的一对纵行隆起,称为尿生殖嵴(urogenital ridge)。尿生殖嵴进一步发育,中部出现一条纵沟,将其分成内、外两部分。外侧部分较长而粗,为中肾嵴(mesonephric ridge);内侧部分较短而细,为生殖腺嵴(gonadal ridge)。
在人胚第5周时,原始生殖细胞通过阿米巴样运动沿着后肠的背侧肠系膜运动并达到发育胚胎中的腰部区域,即在将来形成生殖嵴的区域。在中肾体(或沃尔夫体)的前内侧排列的体腔上皮增厚形成了生殖嵴,并为性腺的支持细胞提供营养;如果原始生殖细胞无法到达生殖嵴部,则性腺无法发育,在女性中即会发生常见的性腺发育不全综合征。在人胚第6周,原始生殖细胞侵入生殖嵴,并被吸纳进入初级性索,从体腔上皮增殖并生长到下面的间充质中,形成性索的主要部分。此时的生殖嵴被称为“未分化的”性腺,因为这一时期在男胎和女胎中的性腺具有相同的外观。此时未分化的性腺是由外部的皮质和内部的髓质组成:在具有XX性染色体复合物的胚胎中,皮质形成卵巢,髓质退化;在具有XY染色体复合物的胚胎中,髓质分化成睾丸,但皮质退化。性索最终成为男性的生精小管及女性的髓质索。初级性索继续活跃增殖,于间充质深处相吻合,并形成一个复杂的网状体,其被视为位于中肾(沃尔夫)体前内侧上的体腔上皮下的隆起。
性腺发育起始于双潜能性腺(bipotential gonad)的形成,继而分化成为成熟的睾丸或卵巢。这一过程依赖于睾丸特异性或卵巢特异性途径的激活,与此同时,相反的途径被持续性地抑制。转录因子调控网络严格调控不同途径的起始和维持,破坏这些网络可以导致人类的性腺发育障碍,在小鼠中则会引起雌雄性别的逆转。
与性腺发育过程相关的基因可以大致分为三类:①形成尚未分化性腺的基因,如 Sf1 、 Wt1 ;②决定性腺可以分化为雄性或雌性的基因,如 Sry 、 Sox9 和 Dax1 ;③促进分化成为雌性或雄性结构的基因,如 Sf1 、 Wt1 和 Wnt4 等。 Sry 仅仅在发育中的性腺表达,而其余基因在发育过程中的表达则不局限于性腺中。
在小鼠的胚胎发育中,双潜能性腺最开始出现于胚胎第10.5天(E10.5)。一些在胚胎发育期间对于未分化的双潜能性腺形成起关键作用的转录因子总结于表2-1。编码这些转录因子的基因的突变可导致性腺发育缺失功能,从而被纤维组织取代。
Emx2 编码同源域转录因子,是果蝇中 emx 基因的同源物,在胚胎早期的原始性腺嵴中表达。 Emx2 基因敲除的小鼠完全缺乏性腺、肾脏和生殖道,证实Emx2蛋白在早期泌尿生殖系统和双潜能性腺的发育中起到关键作用。在发育中的生殖系统中,它在上皮组织中表达且可以被HOXA10负性调节,选择性地剪切,导致产生编码不同蛋白质的多种转录产物变体。人类的 Emx - 2 基因定位于10q26.1,在人类的研究中主要集中在三种组织的表达:端脑背侧、嗅神经上皮和泌尿生殖系统。 Emx - 2 的杂合突变会导致人脑裂畸形,目前尚未发现有早期性腺发育不全的例证。
表2-1 与双潜能性腺发育相关的基因
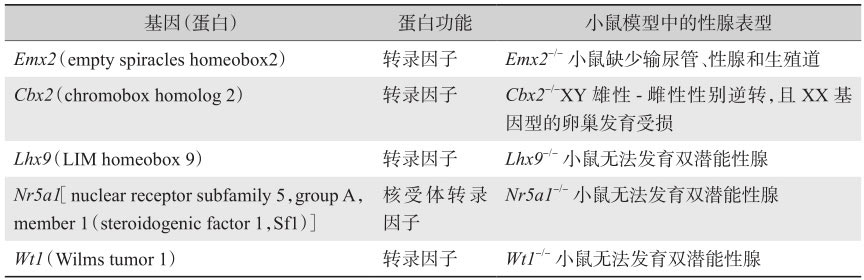
Lhx9 -/- 敲除的小鼠也同样无法发育性腺,并且基因型为XY的小鼠由于缺乏睾酮和AMH,从而在表型上发育为雌性小鼠。此外, Nr5a1 基因是非常早期的性腺发育所需的关键基因,由于 Lhx9 调控 Nr5a1 基因的表达,且小鼠中该基因的缺失会导致性腺发育不全和SF-1表达减低,因此推断LHX9蛋白可能位于转录因子调控网络的较上游位置。
编码类固醇生成因子1(steroidogenic factor 1,SF-1),一种在性腺和所有原发性类固醇生成组织(包括肾上腺)中表达的转录因子。在小鼠中敲除 Nr5a1 基因,会造成缺乏性腺和肾上腺的表型,表明该基因在性腺发育和类固醇生成中有重要作用。此外,Nr5a1在上调睾丸基因 Sox9 的表达中也起到关键的作用。
AMH(由 Amh 基因编码)是睾丸最早产生的激素之一,并且负责雌性米勒管的退化。 Amh 的转录是由Nr5a1和Wt1负责调控的,为早期性腺发育和随后的睾丸分化所必需。与早期性腺和泌尿生殖系统发育期间表达的其他转录因子一样, Wt - 1 基因敲除的小鼠无法发育出性腺和肾脏,证实了其在非常早期的双潜能性腺中具有重要作用。Wt-1包括2个亚型,即含有氨基酸KTS(赖氨酸-苏氨酸-丝氨酸)的+KTS型和不含KTS的-KTS型。+KTS型的缺失会减少 Sry 和 Sox9 的表达,导致46,XY的个体发生性别逆转,-KTS亚型的缺失则会导致性腺形成过程中细胞死亡的增加,从而使性腺呈条索状。
Cbx2 -/- 小鼠性腺发育迟缓并发生雄-雌性别逆转、生殖细胞丢失,导致卵巢体积缩小并且不育,暗示CBX2在卵巢发育过程中发挥了潜在的作用。此外, Cbx2 也参与了几种睾丸基因的上调,包括 Nr5a1 、 Wt1 和 Sry 。最近研究者认为其在人类性腺发育过程中扮演着重要作用,CBX2及其小鼠同源蛋白M33是Polycomb(PcG)蛋白质家族的成员。PcG蛋白是高度保守的转录调节因子,可形成较大分子量的蛋白质复合物,通过调节高级染色质结构发挥其功能。Edi等研究表明,CBX2对于人类性腺发育中卵巢命运的决定及卵巢的维持有着重要的作用。CBX2除了上调雄性相关基因( SOX2 、 SOX3 、 FGF2 、 INSL3 以及 SF1 和 SRY 之外),还可以负性调节雌性相关基因,例如 FZD1 、 PBX1 和 FOXL2 等。作为卵巢发育过程中的抑制因子,CBX2可能是在卵巢中的调控网络的信号通路中精细调控因子之一(其他还包括WNT4和RSPO1),可以防止卵巢非控制性地生长以及最终导致卵巢肿瘤的发生。
生殖腺或性腺,睾丸及卵巢由两种类型的细胞组成,即原始生殖细胞(primordial germ cells,PGCs)及营养支持细胞(卵巢的滤泡细胞和睾丸的支持细胞)。原始生殖细胞是在发育期间建立的第一个生殖细胞群,是卵母细胞和精原细胞的直接前体细胞。在哺乳动物胚胎原肠胚时期,外胚层细胞通过多种复杂信号诱导而成为原始生殖细胞。原始生殖细胞是较大的球形细胞群,直径为25~30mm,具有颗粒状细胞质。生殖细胞的发育通过基因组功能的遗传及表观遗传调控产生了细胞的全能性。
在小鼠胚胎E7.25时,PGCs在初期尿囊的基底部成为可鉴别的40个左右的细胞团,它们在E7.75迁移至正在发育中的后肠内胚层,至E9.5到达肠系膜,而后在E10.5定位于生殖嵴。在上述的PGCs增殖的过程中发生了一个重要的生物学事件,即表观重编程。其中,最重要的是包括基因组印记擦除在内的全基因组去甲基化的过程。
在胚胎E7.25对PGCs进行的基因表达的单细胞分析鉴定出两个基因,即 Fragilis 和 Stella ,它们分别高度和特异性地在PGCs中表达。进一步对PGCs转录组的筛选鉴定出了两个在PGC特化过程中的关键因子: Blimp1 和 Prdm14 。
1) Fragilis :
小鼠干扰素诱导的蛋白样基因1或干扰素诱导的跨膜蛋白3(interferoninduced transmembrane protein 3,IFITM3)是干扰素可诱导的跨膜蛋白家族之一。它在E6.25~E6.5时在最近端外胚层细胞周围开始出现表达,并且其表达在胚外中胚层后部增强。其中碱性磷酸酶(alkaline phosphatase,AP)阳性的PGCs出现在E7.0~E7.25。
2) Stella :
发育多能性相关因子3(developmental pluripotency associated 3,Dppa3)是一类小的高度保守的细胞核-细胞质穿梭蛋白。在E7.0~E7.25, Stella 开始在胚外中胚层表达 Fragilis 的细胞中特异性表达,并持续在迁移的PGCs中表达。 Stella 阳性表达的细胞显示出组织非特异性碱性磷酸酶基因(alkaline phosphatase, ALPL )的高表达。具有 Stella 阳性和 Fragilis 高表达的细胞抑制同源盒基因如 Hoxb1 和 Hoxa1 的表达,然而 Fragilis 阳性但是 Stella 阴性的细胞则保留 Hox 基因的表达。因此可以推测, Stella 阳性的、 Hox 阴性的细胞是已经建立起的PGCs。 Stella 是在受精卵中保护母本基因组和父本印记基因不被全基因组甲基化的关键性因素。
3) Blimp1 和 Prdm14 :
BLIMP1和PRDM14是高度保守的蛋白,与 Tcfap2c ( AP2γ )一起,是在哺乳动物中PGC特化过程中所必需的因子。
A. Blimp1 :促B淋巴细胞成熟蛋白1(B-lymphocyte-induced maturation protein 1, Blimp1 )。BLIMP1阳性的细胞最初出现在胚胎后部最近端的外胚层细胞中,它们数量增多并形成具有强碱性磷酸酶的活性细胞团,并对 Stella 和 Hox 基因的表达具有抑制作用。BLIMP1是一个转录抑制因子,在其氨基末端带有PR(PRDIBF1和RIZ)结构域,在其羧基末端带有5个Krüppel型锌指结构。PR结构域在结构上类似于SET(hairless抑制子,zeste增强子和Trithorax)结构域,显示出组蛋白甲基转移酶活性。BLIMP1的PR结构域未显示出酶的活性。BLIMP1与许多不同表观调控因子相互作用,包括HDAC、Groucho和G9A等。在 Blimp1 敲除的胚胎中,AP阳性的PGCs样的细胞出现在E/MB期,但是其数目减少,未表现出迁移的表型,且无法表现出PGCs特异性的基因。
B. Prdm14 :PR域结合蛋白14(PR domain-containing protein 14,Prdm14)最初于E6.5在BLIMP1阳性的细胞中表达并最终在PGCs中表达。所有BLIMP1阳性的PGCs前体细胞最开始表达 Hox 基因并抑制 Sox2 的表达。尽管如此,从E6.75开始,BLIMP1阳性细胞开始抑制 Hox 基因的表达并开始表达 Sox2 、 Stella 和 Nanog 。此外, Prdm14 在原始细胞中的表达依赖BMP4信号通路。 Prmd14 的缺失导致PRDM1阳性细胞减少,对 Prdm1 、 Prdm14 基因敲除的小鼠胚胎的研究发现,这两种蛋白彼此独立发挥作用。
综上所述,PGCs承担了三个关键的生物学事件:①体细胞中胚层的程序性抑制;②重新获得潜在的多能性;③随后的表观重编程。在小鼠甚至所有哺乳动物中的早期原肠胚形成期间生殖细胞系在一系列信号分子的刺激下,最终分化为PGCs。
在20世纪70年代左右,科学家发现在脊椎动物的胚胎发育过程中,存在原始生殖细胞能够精确地迁移到目的地的现象。而人类原始生殖细胞直到在E21才可辨别,并在尿囊起源附近的卵黄囊壁中的内胚层细胞中可见。因此,原始生殖细胞在距离生殖嵴的最终确定位置有一定的距离,需要通过迁徙及精确地调控才能使得原始生殖细胞到达生殖嵴。驱使原始生殖细胞进行迁移的因素及其作用方式都是学者研究的方向。近年来,传统分子生物学手段及高通量测序技术的发展逐步揭开了原始生殖细胞迁移机制的面纱。
在迁移之前,原始生殖细胞开始具有运动活力,并会接收到特定信号的引导。在不同物种中,PGCs迁移受到不同信号通路、转录因子及细胞极性等机制的调控。
在果蝇的研究中,PGCs特化之后展示出了迁移的特性,在原肠胚形成时期,PGCs通过组织运动进入后形成胚胎的中肠袋。在后中肠中,PGCs在腔中彼此形成紧密簇,但与周围的体细胞几乎没有接触。该PGCs簇具有径向结构,每个细胞的前缘朝向中肠后部的特征。随后,PGCs开始向周围的后中肠细胞延伸细胞突起,并且彼此失去黏附。当细胞从PGCs簇中分散并通过后中肠单独迁移时,具有活性的PGCs迁移开始。
小鼠的PGCs最初在后原条中可以鉴定出来,随后PGCs开始展示出极性的形态学,并延伸出细胞质突起,其通过原条进入相邻的后胚胎内胚层,胚外外胚层和尿囊内膜。小鼠PGCs的迁移大约经历4天,整个迁移过程包括早期被动的迁移过程和晚期主动的迁移过程。第一阶段(E7.5~E9.0),从原条后部及尿囊根部内胚层来源的PGCs被卷入胚胎内部进入后肠,并到达后肠内胚层,此过程为PGCs随内陷的内胚层被动迁移的过程。第二阶段(E9.5~E11.5),PGCs离开后肠内胚层,沿着背肠系膜,最终迁移到生殖嵴,此为主动迁移的过程。PGCs表现出迁移细胞的超微结构,如伪足等结构,以阿米巴运动方式主动向生殖嵴迁移,其末端附着在细胞外基质或周围的体细胞上。
1)迁移的启动:
干扰素诱导的跨膜蛋白1(interferon-induced transmembrane protein 1,IFITM1):IFITM1介导小鼠PGCs迁移的起始。该蛋白为膜表达蛋白,可以参与多种细胞学过程,包括细胞黏附等。RNA干扰技术将原条中的 Ifitm1 进行敲减可导致PGCs无法进入内胚层,因此认为IFITM1驱使PGCs从中胚层进入至内胚层。然而,最新的研究表明,在胚胎中敲除 Ifitm1 并不影响PGCs迁移的启动,因此这一蛋白在PGCs迁移启动过程中的作用及其作用机制仍存在争议。
2)迁移的路径:
PGCs的迁移路径必须经过精细的调控才能引导PGCs通过发育的胚胎向体细胞性腺前体(somatic gonadal precursors,SGPs)迁移。在小鼠胚胎E7.5时,PGCs启动迁移机制,细胞从后原条移动至内胚层。随后在E8~E9.5时,小鼠的PGCs在后肠中向前延伸,其路径与果蝇非常相似(在果蝇中,PGCs从后肠组织移动至中胚层)。紧接着在E10.5~E11.5期间,PGCs双向迁移至生殖嵴并形成性腺。
1) SOX17 :
性别决定区域Y盒17(sex determining region Y box 17, SOX17 )是编码参与胚胎发育调控和决定细胞命运的 SOX 转录因子家族的成员。编码的蛋白质和其他蛋白形成复合物可作为转录调节因子。在敲除 Sox17 转录因子的小鼠中,后肠的内胚层延伸受到抑制,PGCs无法正确地迁移至生殖嵴,从而使PGCs分散在胚外内胚层。
2) SDF1/CXCR4 :
基质细胞来源因子1(stroma-derived factor 1, SDF1 )和趋化因子受体4(C-X-C motif chemokine receptor, CXCR4 )作为PGCs的诱导系统,该信号通路对于细胞迁移出内胚层并不具有一定的功能,但对于PGCs迁移至生殖嵴终末阶段是必需的。 SDF1 表达于生殖嵴和周围的间充质,而 CXCR4 表达于PGCs。CXC4R4b/SDF-1a信号途径受到损害,PGCs不能正常定向迁移,且遍布整个胚胎。 SDF1 或 CXCR4 的敲除导致仅极少量的PGCs到达生殖嵴,而异位表达 SDF1 将引起PGCs迁移至新的位点。
3)c-kit/steel系统与PGCs的迁移:
受体酪氨酸激酶(KIT proto-oncogene receptor tyrosine kinase,c-kit)与其配体steel(或KITlG)很久以来被认为在PGCs增殖迁移和生存中起到了重要的作用。c-kit-steel相互作用对于PGCs沿后肠的内胚层的迁移过程发挥了重要作用。近期的科学研究已经表明了这些因子特定的迁移作用,steel和c-kit调控PGCs的一般运动能力,steel功能的缺失导致PGCs在正确的方向上进行迁移,但PGCs在数量上明显下降。在果蝇中,该表型的PGCs功能的缺失被认为与JAK-STAT通路的干扰有关。
黏附分子:除了信号通路,也有证据表明黏附分子在小鼠PGCs迁移的过程中具有一定作用。E-cadherin随PGCs迁移出后肠时表达。干扰E-cadherin的功能会影响PGC-PGC之间的相互作用,进而导致PGC遗留在性腺外部。PGCs同样表达整合素β1(β1 integrin),对于PGCs正常地迁移出后肠而进入生殖嵴是必需的。
4)HMGCR:
3-羟基-3-甲基戊二酰辅酶A还原酶(3-hydroxy-3-methylglutaryl-CoA reductase,HMGCR)是胆固醇合成的限速酶。在体外培养系统中抑制HMGCR损害了生殖细胞的迁移。在小鼠中,胆固醇及胆固醇合成中间产物如类异戊二醇,都参与了PGCs的迁移过程。此外,胆固醇被发现在生殖嵴中富集,进一步表明其在PGCs迁移过程中的潜在作用。但是HMGCR通路在体内的作用仍有待发掘,可通过基因靶向敲除以及更深入的基因表达分析来确定。
综上所述,PGCs迁移的启动和迁移路径受到多种因素的影响。探究PGCs的迁移机制,不仅可以了解迁移过程中细胞内信号传导和细胞间信息传递,而且对阐明PGCs迁移异常所致的人类肿瘤的发生机制有重要意义。目前研究多集中于小鼠、果蝇、斑马鱼等动物体内,若要探究人类的迁移机制还需要克服更大的困难,进行更深入细致的科学探究。
虽然胚胎的染色体性别在受精时就已经确定,但直至胚胎发育至6~7周时,生殖腺才开始分化为睾丸或者卵巢。在缺少Y染色体的胚胎中,生殖嵴区域发育缓慢,且最初未分化的雌性性腺在大约人胚第10周才发育成为可识别的卵巢,此时其皮质的特征变得明显。X染色体具有促进卵巢发育的基因,常染色体上的基因也在卵巢的器官发生中起到一定的作用。
在胚胎时期,体腔生殖上皮细胞增殖进入其下的间充质中。性腺索延伸到卵巢中央并形成基本的卵巢网(rete ovarii)。人胚第10周后,初级性索退化,被基质和血管替代,成为卵巢髓质,并且出现表面上皮又一次向深层增殖形成新的细胞索,成为次级性索(secondary sex cords)或皮质索(cortical cord),最终构成了卵巢的皮质。随着皮质索体积的增加,原始生殖细胞逐渐被吸收融入其中,在大约人胚第16周,皮质索开始分裂成为独立的细胞团,其内中央是由原始生殖细胞分化而来的卵原细胞,是一类胞质清晰的大细胞,其周围是一层由次级皮质索或性索细胞分化而来的小而平的卵泡细胞,两者构成始基卵泡。始基卵泡分布在结缔组织基质中,其数目有限,出生时大约有30万~200万个。
在最初的始基卵泡储存库中,大约有400~500个卵泡在青春期和至绝经前发育成熟,并产生可受精的卵子。卵原细胞在胚胎时期发生了活跃的有丝分裂,目前认为人类出生后无卵母细胞的产生。初级卵母细胞不能自我复制,因此出生后卵巢内的初级卵母细胞不再增多。当初级卵母细胞被1~2层立方形或低柱状滤泡细胞包围时,其被称为初级卵泡。大多数卵泡在青春期前一直处于静止状态。出生后,卵巢表面上皮与腹膜间皮连续的单层细胞相连续,表面上皮细胞通过薄层纤维囊,即白膜,与皮质中的卵泡相互分离。
原始性腺索在女性胚胎性腺中并不突出,延伸到髓质中以形成原始的卵巢网。卵巢网和初级性索通常会退化并消失。在性腺未分化时期,性索的第一次增殖并向腺体的中心区域重新定位,形成髓质索。髓质索和卵巢网以及其和中肾之间的连接,退化并最终组成罗森米勒小体(Rosenmüller’s body)或者称为副卵巢。
相比于睾丸,对于卵巢分化发育的相关理论与研究则少得多,目前尚未完全清楚。然而,仍有一些基因被鉴定出来(表2-2),为我们对卵巢的发育调节提供了更多的见解。
表2-2 小鼠中决定性腺发育命运的关键分子
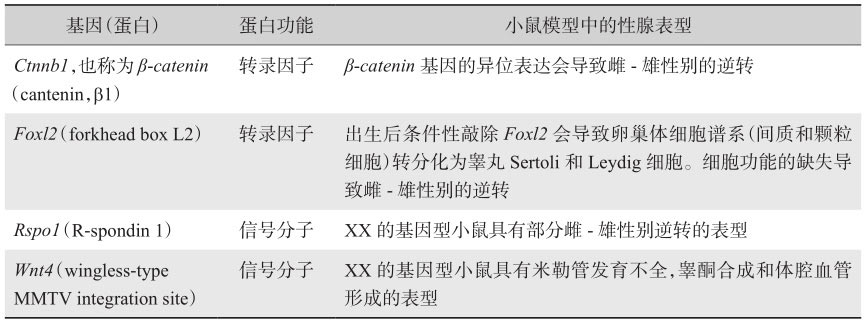
Foxl2 (forkhead box L2)是叉头框基因家族的成员,编码进化中保守的转录因子。在小鼠中, Foxl2 是在发育中的雌性特异性性腺(卵巢)中最早上调的基因之一,表明该基因在早期卵巢分化中的重要功能。在基因型XY的小鼠中 Foxl2 的过表达和XX小鼠中 Foxl2 敲除导致性腺异常发育,但并不造成性别逆转。然而,在山羊模式动物中, Foxl2 缺失突变或 Foxl2 上游11.7kb区域的缺失导致雌性向雄性的性别逆转,进一步支持 Foxl2 在雌性性腺发育过程中的作用。 Foxl2 可以从早期胚胎性腺分化开始至成年后抑制睾丸分化相关的基因的表达,也证实其在出生后卵巢的维持中起到重要作用。
是卵巢特异性激活相关的基因。 Wnt4 和 Rspo1 是WNT信号通路上的两个重要组成部分,在卵巢分化发育过程中具有重要作用。 Wnt4 和 Rspo1 是通过激活 β - catenin 来发挥作用的。 β - catenin 又反过来调节多种基因的转录,其中包括多种重要的卵巢成分,如 Wnt4 和 Fst 。在小鼠胚胎E12.5, Rspo1 和 Wnt4 开始在卵巢内特异性表达。 Rspo1 和 Wnt4 是由卵巢的体细胞表达的,其中,在E12.5和E14.5小鼠胚胎卵巢中的体细胞和生殖细胞的细胞膜中检测到了RSPO1的表达,而在胚胎E12.5 Rspo1 -/- 的性腺中无法检测到WNT4的表达;相反,在 Wnt4 -/- 的性腺中RSPO1可以持续表达,证明了RSPO1/WNT信号通路可以维持WNT4的表达。 Wnt4 的缺失可能会导致前颗粒细胞异常分化。
随着 Rspo1 和 Wnt4 的表达,WNT/β-catenin在E12.5后在卵巢的体细胞和生殖细胞中以性别特异性的方式被激活,如 Axin2 的表达,是该信号通路上通用的激活标志物。在基因型为XY的性腺中,WNT/β-catenin信号通路随着 Sry 的表达而逐渐下调。体外研究同样表明,SRY可以在蛋白水平上与其相互作用来拮抗CTNNB1,从而将CTNNB1靶向至核小体,来触发其降解并抑制CTNNB1介导的转录活性。在XY体细胞中异位激活CTNNB1会通过扰乱睾丸的命运而促进卵巢发育,导致雄-雌性的性别逆转,该研究证实了β-catenin信号通路是雌性性别决定的通路。
哺乳动物的性别决定需要GATA家族的转录因子(GATA4和GATA6)及其辅助因子FOG2(Zfpm2,zinc finger protein,multitype 2)之间的相互作用,而且已经被认为是多种发育过程中的关键驱动因素。Eifmenko等利用转基因敲除鼠证实GATA4-FOG2在卵巢发育和卵泡形成中的重要作用。不同于个体在性别决定期间需要GATA4-FOG2复合体,接下来的卵巢分化过程需要的是GATA4,但是并不依赖FOG2。在卵巢中 Gata4 表达的缺失会损坏颗粒细胞的增殖和间质细胞的募集,同时卵巢皮质的始基卵泡数目减少,导致卵泡无法发育。
此外,GATA4-FOG2复合物的作用还有其可以作为 Dkk1 基因的抑制剂, Dkk1 编码经典β-catenin信号通路上的一个分泌型抑制蛋白,是卵巢发育中GATA4-FOG2抑制的靶标。性腺中 β - catenin 基因的组织特异性的敲除破坏了雌性发育。GATA家族蛋白在性别决定和性腺分化中的作用最近引起研究者的广泛关注,成为了胚胎期性别发育调控作用中较为火热的研究对象。
睾丸和卵巢的分化发育途径之间存在复杂的相互作用。在发现SRY作为睾丸的关键性决定因素后不久,McElreavey等提出“Z模型”的假设。在这种假设下,XX性腺产生了“Z因子”,通过抑制一种或多种促睾丸基因来促进卵巢发育。根据该模型, Sry 或另一早期雄性特异性基因通过阻断Z因子的活性来抑制卵巢发育。尽管尚未有明确的Z因子被鉴定出来,一些研究已经证实雄性和雌性通路在性腺分化期间和功能性睾丸或卵巢完全发育后相互拮抗。
Sox9 在早期睾丸发育的过程中有重要的作用,其表达和调控在特定的卵巢或睾丸形成的通路中占据中心地位。Sekido等发现在小鼠中, Sry 和 Nr5a1 被证实通过睾丸特异性Tesco增强子来上调 Sox9 。在 Sry 表达停止后, Sox9 通过该增强子维持其自身的表达,从而使睾丸分化进行。体外实验表明,雌性特异性转录因子 Foxl2 可以与Tesco结合并抑制其活性,从而阻止发育中的卵巢高水平的 Sox9 表达。Tesco可能与其他调控元件协同作用,通过形成三维环状结构导致 Sox9 表达的起始、上调及自我维持。
Foxl2 的另外一个靶标是 Nr5a1 。Taksawa等发现,通过拮抗Wt1-KTS型, Foxl2 在发育中的卵巢中抑制 Nr5a1 的表达。性腺分化的另外两个相互拮抗的信号分子是 Fgf9 和 Wnt4 。在睾丸发育中, Fgf9 被显著上调并起到维持 Sox9 表达以及下调雌性特异性基因如 Wnt4 的表达的作用,反过来, Sox9 会激活 Fgf9 的表达。在卵巢中,这个反馈环可能是通过 β - catenin 的激活而被 Wnt4 阻断。
在成熟的XY性腺中, Dmrt1 维持睾丸通路并抑制卵巢通路。在出生后的XY小鼠睾丸的支持细胞中敲除 Dmrt1 会诱导这些睾丸支持细胞转分化为颗粒细胞表型:生殖细胞开始具备雌性化特征,并且性腺开始产生雌激素。此外, Foxl2 也可以被诱导表达,这也提示为雌性途径的激活。另有研究表明, Dmrt1 和 Sox9 是通过抑制雌性性别决定的基因(如 Foxl2 )来保持出生后睾丸的维持。 Dmrt1 也是抑制睾丸中过量视黄酸信号传导所必需的,其作用是进一步阻断雌性性别决定基因的表达。
综上所述,在过去的十几年研究中,随着对调控 Sry 表达的新基因的鉴定,我们对性腺特化的调控与之后性别分化的认识已逐渐深入。除此之外,表观遗传修饰和不同种类的microRNA也已被发现作为支持性腺发育的新的调控层。尽管如此,目前仍缺乏对功能完好的睾丸和卵巢的调控网络的全面概括。若期待对调控性腺分化复杂网络进行全面的了解,必须在基因、表观遗传以及其他等多个层面进行探索。
卵母细胞的形成过程又被称作卵子发生(oogenesis)。在胚胎期会形成初级卵母细胞,而后在排卵的过程中会形成次级卵母细胞。
表2-3 卵子发生过程中的细胞种类及特性
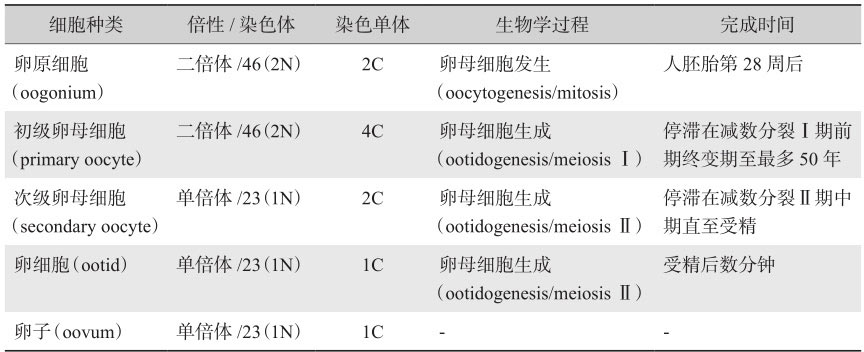
在哺乳动物中,卵子发生起源于胚胎卵巢中PGCs向卵母细胞的分化。其包括几个亚过程(表2-3):卵母细胞发生(oocytogenesis),卵母细胞生成(ootidogenesis),以及最后成熟卵子的形成(卵子形成本身)。卵泡发生(folliculogenesis)是一个独立的子过程,伴随并支持上述三个子过程。PGCs经历有丝分裂,形成了卵原细胞,卵原细胞保持有丝分裂的状态,随后进入减数第一次分裂,历经细线期(leptotene)、偶线期(zygotene)、粗线期(pachytene),最后静止于双线期(diplotene),形成初级卵母细胞。进入青春期后,每次月经周期中仅招募少量的初级卵母细胞,且仅形成一个成熟的卵子。初级卵母细胞于排卵前完成减数第一次分裂,产生单倍体次级卵母细胞并排出第一极体。次级卵母细胞进入减数第二次分裂并停滞于中期,若发生受精,则完成减数分裂过程,排出第二极体。与卵子的发生同步,卵泡从始基卵泡发育成排卵前卵泡。
始基卵泡由卵巢内单层扁平前颗粒细胞围绕初级卵母细胞形成,是卵巢储备的唯一形式,也是女性生殖的基本单位。从胚胎期16周开始到出生后6个月,始基卵泡逐渐形成,随着始基卵泡池的建立,始基卵泡就开始执行启动及凋亡过程。始基卵泡的募集分为初始募集和周期募集。初始募集为非促性腺激素依赖性的,始基卵泡在卵巢内因子及其他调控因素下脱离始基卵泡池,而向初级卵泡转化的过程。分为两个阶段:第一阶段为前颗粒细胞由扁平状向立方状转变,并伴随颗粒细胞的增殖;第二阶段为卵母细胞体积增大和颗粒细胞继续增殖。周期募集是指在每个月经周期中,当促性腺激素周期性变化,对这种变化发生应答的卵泡逐步转变为次级卵泡。始基卵泡的募集过程在卵巢生物学上有很重要的意义,直接影响了女性一生中能提供的卵子数量。
综上所述,卵巢发生的整体过程可概括为:人胚第4周时,尿囊处有许多源于内胚层的圆形的原始生殖细胞,于第6周经背侧肠系膜陆续向生殖嵴迁移,进入初级性索内。人胚第5周时,中肾嵴形成生殖腺嵴,其逐渐形成许多细胞索,称为初级性索。若体细胞和原始生殖细胞无 Sry 表达,则未分化性腺向卵巢方向分化。人胚约第10周后初级性索向深部生长,在该处形成不完善的卵巢网。随后,初级性索与卵巢网都退化,被血管和基质所替代,形成卵巢髓质。此后,生殖腺表面上又形成新的细胞索,称之为皮质索,它们较短,分散于皮质内。约在人胚第16周时,皮质索断裂形成许多孤立的细胞团,即为始基卵泡。胚胎期生殖细胞及生殖器官的发育受到多种基因及转录因子和信号通路的共同精密调控,在后续卵巢正常发育过程起到了关键作用。在过去的十几年里,随着对调控 Sry 基因表达的新基因的发现,科学家对性腺调控及随后的性别分化的理解不断加深。此外,支持性腺发育的新的调控层面(如表观遗传修饰和非编码RNA等)已经被发现。然而,对一个功能完备的卵巢分化过程中所需要的调控网络,仍然缺乏全面的了解。如果需要更加完善地理解调控性腺分化的复杂网络,未来有待于利用科技最新进展在表观遗传、非编码RNA等方向进行研究,以更深入、更全面地阐释性腺特别是卵巢分化的关键点及调控网络。
(马菱蔚)
1.Eggers S, Ohnesorg T, Sinclair A.Genetic regulation of mammalian gonad development. Nature Reviews Endocrinology, 2014, 10: 673-683.
2.Sekido R, Lovell-Badge R.Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature, 2008, 453: 930.
3.Eid W, Opitz L, Biason-Lauber A.Genome-wide identification of CBX2 targets: insights in the human sex development network. Molecular Endocrinology, 2015, 29: 247-257.
4.Saitou M, Kagiwada S, Kurimoto K.Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development, 2012, 139: 15-31.
5.Rea S, Eisenhaber F, O’Carroll D, et al.Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 2000, 406: 593.
6.Ohinata Y, Ohta H, Shigeta M, et al.A signaling principle for the specification of the germ cell lineage in mice. Cell, 2009, 137: 571-584.
7.Yamaji M, Seki Y, Kurimoto K, et al.Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nature genetics, 2008, 40: 1016.
8.Moore KL, Persaud TVN, Torchia MG.Before we are born: essentials of embryology and birth defects. 9th ed. Philadelphia: Elsevier Health Sciences, 2015.
9.Richardson BE, Lehmann R.Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nature reviews Molecular Cell Biology, 2010, 11: 37.
10.Hara K, Kanai-Azuma M, Uemura M, et al.Evidence for crucial role of hindgut expansion in directing proper migration of primordial germ cells in mouse early embryogenesis. Developmental Biology, 2009, 330: 427-439.
11.Ara T, Nakamura Y, Egawa T, et al.Impaired colonization of the gonads by primordial germ cells in mice lacking a chemokine, stromal cell-derived factor-1 (SDF-1). Proceedings of the National Academy of Sciences, 2003, 100: 5319-5323.
12.Molyneaux KA, Zinszner H, Kunwar PS, et al.The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development, 2003, 130: 4279-4286.
13.Boulanger L, Pannetier M, Gall L, et al.FOXL2 is a female sex-determining gene in the goat. Current Biology, 2014, 24: 404-408.
14.Efimenko E, Padua MB, Manuylov NL, et al.The transcription factor GATA4 is required for follicular development and normal ovarian function. Developmental Biology, 2013, 381: 144-158.
15.McElreavey K, Vilain E, Abbas N, et al.A regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male development. Proceedings of the National Academy of Sciences, 1993, 90: 3368-3372.
16.Minkina A, Matson CK, Lindeman RE, et al.DMRT1 protects male gonadal cells from retinoid-dependent sexual transdifferentiation. Developmental Cell, 2014, 29: 511-520.
17.Matson CK, Murphy MW, Sarver AL, et al.DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature, 2011, 476: 101.
18.Mead TJ, Wang Q, Bhattaram P, et al.A far-upstream (-70kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic Acids Research, 2013, 41: 4459-4469.
19.Takasawa K, Kashimada K, Pelosi E, et al.FOXL2 transcriptionally represses Sf1 expression by antagonizing WT1 during ovarian development in mice. The FASEB Journal, 2014, 28: 2020-2028.
卵巢从形成后即进入缓慢的生长过程,并在此过程中由髂窝下缘缓慢下降至盆腔,体积逐渐增大。卵巢的生长过程伴随着卵泡的发生过程,始基卵泡持续激活,卵泡数目不断减少,但由于早期缺乏下丘脑、垂体激素的支持,卵泡只能发育到窦前卵泡阶段,少数能发育至排卵前卵泡,但都最终闭锁。此时的卵巢具有一定的激素分泌功能,但水平较低。卵巢由多种细胞成分组成,不同细胞成分的相互作用在卵泡的发育成熟和卵巢的成长过程中发挥重要作用。
卵泡是一个由卵母细胞和其周围的颗粒细胞及卵泡膜细胞共同组成的多细胞功能单元,是卵巢的生命源泉,卵泡的数量和质量直接影响着卵巢的生命力。卵泡中的卵母细胞与周围细胞通过细胞间连接运输代谢产物,并通过一系列细胞因子以旁分泌及自分泌的方式相互调节,主要包括TGF-β家族成员。卵巢中的其他成分对卵泡的存活和生长同样重要,其中卵巢基质为生长卵泡中产生的生长因子和调节因子提供屏障。此外,卵巢上皮细胞、免疫细胞及神经调节因素也在卵巢中发挥重要作用。
卵母细胞是罕见的巨大细胞,其细胞核较大,且具有高转录活性,也被称为“生发泡”。卵母细胞的细胞质含有丰富的细胞器以及蛋白质。在卵泡的生长过程中,卵母细胞的关键任务是产生供排卵、受精以及积蓄着床前胚胎形成新基因并开始转录所需要的所有成分。卵母细胞中基本的细胞质成分包括核糖体、线粒体和母体mRNA,这些成分在卵母细胞生长的过程中不断累积,为卵母细胞成熟和胚胎发育提供必需的蛋白质。由角蛋白、微管蛋白和其他高度丰富的卵母细胞蛋白组成的细胞质中含有丰富的核糖体和母体mRNA。卵母细胞与体细胞线粒体在结构和DNA含量方面具有显著不同。体细胞线粒体是球形的,几乎没有嵴,并且每个细胞只含有1~2个线粒体DNA。卵母细胞含有比体细胞更多的线粒体。在形成原始生殖细胞时大约含有10个线粒体,而每个原始卵母细胞含有高达6 000个线粒体,在生殖细胞迁移到性腺、进入减数分裂和始基卵泡形成的整个过程中,线粒体的数量迅速增加。随着卵母细胞生长的开始,线粒体继续复制,在完全成熟的人卵母细胞中估计有300 000~400 000个线粒体。
卵母细胞特异性基因在卵泡发育成熟、受精及着床前的发育等过程中发挥关键作用。其中 Figla 、 Sohlh1 和 Lxh8 的表达是裸卵母细胞形成卵泡的必要条件,而 Nobox 是始基卵泡募集为初级卵泡的关键基因。 Dazl 、 Cpeb1 和 Ybx2 (以前称为 Msy2 )是调节卵母细胞内mRNA表达的DNA或RNA结合蛋白。此外,卵母细胞外基质和透明带(zona pellucida,ZP)的形成伴随着卵母细胞的发育过程。
透明带对发育中的卵母细胞、输卵管中的排卵卵泡以及卵裂期的胚胎均发挥重要的保护作用。透明带保护卵泡中正在发育的卵母细胞、输卵管中的排卵卵泡以及卵裂期胚胎。它作为与精子接触的初始部位,在受精后,成为阻止多精子穿透的屏障。 Zp1 ( Zpa )、 Zp2 ( Zpb )和 Zp3 ( Zpc )是编码透明带主要硫酸化糖蛋白的三个基因。ZP2和ZP3蛋白聚合物形成的花环体被ZP1蛋白贯穿连接,形成透明带的亚单元。 Figla 调节卵母细胞生长过程中上述基因的协调表达。研究显示,人类和大鼠有第四个ZP1样亚基(ZP4),而小鼠没有。多年来ZP3被认为是小鼠中主要的精子受体,ZP2是次要的受体。然而,现有的证据表明识别ZP2氨基末端的特定区域是小鼠和人的精子成功穿透透明带的必需条件。受精后,从卵子中释放出名为卵黄素的皮质颗粒金属蛋白酶可切割ZP2,阻止其余的精子与ZP结合,以防止多精子受精。缺乏ZP1的小鼠表现为透明带的结构异常和繁殖力的降低;缺乏ZP2的小鼠表现为薄型透明带,缺乏排卵前卵泡,繁殖期小鼠卵巢中窦卵泡数量显著减少,排卵减少并且不能形成两细胞期胚胎。而且对ZP2缺乏的雌性小鼠的卵母细胞进行体外受精,形成的囊胚不能正常发育。缺乏ZP3的小鼠虽然其他透明带蛋白均可正常表达,但不能形成透明带,也几乎不能排卵,且缺乏生育能力。与 ZP2 突变小鼠一样, ZP3 缺陷小鼠的体外受精卵发育不会超过囊胚期。
此外,通过小鼠模型鉴定出了一系列卵母细胞特异性的“母体效应”基因,这些基因在卵母细胞中表达,但仅在着床前胚胎发育过程中发挥作用。其中 Nlrp5 、 Khdc3 、 Ooep 和 Tle6 基因编码的蛋白质在卵母细胞中形成皮层下母源复合体(subcortical maternal complex,SCMC)。此外,基于其在卵母细胞和卵裂期胚胎中的相似定位模式,肽酰基天冬氨酸二硫异构酶Ⅵ型(PADI6)也可在该复合体中发挥作用。缺乏 Nrlp5 、 Padi6 或 Ooep 的小鼠卵母细胞没有细胞质网格(cytoplasmic lattices,CPLs),合成蛋白质的能力缺陷,且受精后不能发育。KHDC3调节卵母细胞和早期胚胎的纺锤体功能; Khdc3 敲除的雌性小鼠的胚胎由于非整倍体的发生率高而发育不良。与 Nlrp5 相关的基因 Nlrp2 在卵泡发育过程中的卵母细胞和颗粒细胞中均有表达,也是早期胚胎发育成功所必需的母体蛋白编码基因。小鼠卵母皮层下母源复合体基因与人卵母细胞具有相同的表达模式,且功能相似。 TLE6 磷酸化位点纯合点突变的女性通常由于受精后胚胎分裂失败而不育。同样,缺乏PADI6的人卵母细胞受精后发育停滞。随着研究的不断深入,逐渐发现了一系列“母体效应基因”,如 Zar1 (zygote arrest 1)编码的细胞质蛋白决定受精卵向裂解胚胎转变过程,但机制尚不明确; Gclm (glutamate cysteine ligase,modifier subunit)编码调节谷胱甘肽合成的蛋白,是控制细胞氧化还原状态的关键成分,缺乏GCLM的小鼠胚胎不能发育成囊胚; Npm2 (nucleoplasmin 2)是卵母细胞成熟前编码产生的一种核蛋白,影响异染色体的重组和组蛋白去乙酰化,缺乏NPM2的卵子可以正常排卵和受精,但往往无法完成着床前胚胎发育; Dppa3 除了在原始生殖细胞中发现的作用外,也是着床前胚胎正常发育所需的母体效应基因。
一系列的证据表明卵母细胞决定卵泡的发育进程。卵母细胞对卵泡生长的调控作用主要是通过由卵母细胞产生的卵母细胞选择性或特异性TGF-β超家族成员例如生长分化因子-9(growth differentiation factor-9,GDF-9)和骨形态发生蛋白-15(bone morphogenetic protein-15,BMP-15)介导。通过对小鼠的基因干预和绵羊中编码GDF-9和BMP-15基因自发突变体表型的观察,可明确GDF-9和BMP-15在卵泡发育过程中的重要作用。在人卵母细胞中亦发现高表达的GDF-9和BMP-15与随后卵母细胞和胚胎的整体质量密切相关。
GDF-9由5q31.1染色体上的基因编码,在卵母细胞和灵长类的颗粒细胞中高表达。GDF-9缺陷小鼠的卵泡生长停滞在初级阶段,但卵母细胞以比野生型卵母细胞更快的速度继续发育,并发展成为正常小鼠的窦卵泡时期的卵母细胞。然而,由于颗粒细胞与卵母细胞之间的相互连接的超微结构的异常,最终卵母细胞死亡,仅留下一条透明带。与此同时,GDF-9缺陷小鼠卵泡周围不形成卵泡膜,提示GDF-9参与卵泡膜的组成或细胞增殖的调控。相反地,在大鼠中的研究发现,添加GDF-9可刺激初级卵泡的生长,与缺乏GDF-9的小鼠在初级阶段的阻滞相一致。GDF-9通过与激活素样受体(ALK)-5(TGF-βRI)和2型BMP受体(BMPR-2)受体复合物的相互作用,对颗粒细胞和卵泡膜细胞发挥调控作用,并且具有显著的种属特异性。在啮齿动物中,GDF-9刺激颗粒细胞分化,包括诱导黄体生成素(luteinizing hormone,LH)受体和甾体生成。在卵丘细胞中,GDF-9促进透明质酸合成酶2、五肽3和肿瘤坏死因子诱导基因6( TSG - 6 )的表达,后者转录的蛋白质参与卵丘复合体的蛋白多糖细胞外基质的形成。GDF-9刺激COX-2和前列腺素的合成和孕酮的分泌,同时还抑制尿激酶的表达,并抑制卵丘细胞LH受体的表达,阻止卵丘细胞黄体化。暴露于最高浓度的GDF-9有助于卵母细胞周围颗粒细胞的独特表型的形成。GDF-9在体外刺激人卵泡膜细胞增殖,抑制其类固醇生成,与小鼠卵巢中GDF-9在调控卵泡膜发育的作用模式一致。
也称为GDF-9b,由X染色体上的基因编码,是卵母细胞产生的TGF-β超家族的另一重要成员。它在结构上与GDF-9相似,并且具有相似的表达模式。小鼠 Bmp15 基因的靶向性敲除导致卵巢形态异常、排卵和受精率显著降低,从而导致其生育力显著下降。 Bmp15 和 Gdf9 突变的杂合子小鼠由于卵泡生成和卵丘细胞功能异常导致生育能力严重受损。然而,绵羊 Bmp15 基因的自发性突变体(如Inverdale和Hanna绵羊)的表现与 Bmp15 基因敲除的小鼠完全不同。在杂合状态下,卵泡排卵数量反而增加,从而增加了繁殖力。但在纯合性突变的母羊中观察到与 Gdf9 敲除的小鼠相似的原发性卵巢衰竭表型。在体外,BMP-15促进颗粒细胞有丝分裂。因此,体内BMP-15的缺乏可能导致与纯合突变绵羊体内类似的卵泡发育障碍。BMP-15的受体是由BMPR1B(ALK6)和BMPRII组成的受体复合物。根据突变等位基因拷贝数的不同,Booroola绵羊BMPR-1B的点突变与排卵率的增加有关。小鼠中 Bmpr2 基因的靶向性缺失不影响卵泡发育,却因为卵丘细胞膨胀的缺陷阻止了体内受精导致小鼠不孕。
BMP-15和kit配体之间以负反馈方式相互作用:BMP-15刺激颗粒细胞kit配体的表达,而kit配体抑制BMP-15在卵母细胞中的表达。在卵母细胞的存在下,BMP-15和kit配体促进颗粒细胞的有丝分裂。而kit(kit配体受体)只在卵母细胞表达,并且kit配体抑制卵母细胞BMP-15(颗粒细胞有丝分裂原)的表达,这表明卵母细胞可能产生其他颗粒细胞有丝分裂原。
GDF-9和BMP-15先以二聚体的前体蛋白合成,然后经蛋白水解以产生生物活性分子。值得注意的是,使BMP-15失活的Inverdale绵羊突变极大地损害了突变型BMP-15和野生型GDF-9在共表达细胞内的蛋白质水解过程。因此Inverdale羊表型发生的原因至少一部分是由于突变型BMP-15对野生型GDF-9再修饰的干扰而引起的GDF-9功能的缺乏。相同地,人共表达细胞BMP-15和GDF-9突变可能导致翻译后加工受损,降低功能蛋白的产生,从而导致相关的卵巢早衰的发生。研究发现,不仅GDF-9和BMP-15的同源二聚体具有生物活性。这两种蛋白的异源二聚体在调节颗粒细胞存活、颗粒细胞支持卵母细胞代谢的功能、成熟过程中卵丘细胞的扩张等方面都具有非常强的生物活性。因此,GDF-9:BMP-15异二聚体可能才是卵母细胞分泌的必需功能配体。
颗粒细胞起源于卵巢表面上皮,具有两种形成波。第一波参与卵巢髓质卵泡的形成,第二波参与卵巢皮质中卵泡的形成。颗粒细胞的形成是由GATA结合蛋白4(GATA4)表达细胞驱动的。GATA4、WNT4、R-spondin 1(RSPO1)、β-catenin和FOXL2协同促进胎儿期颗粒细胞的发生并调节卵泡生成。包绕每个卵母细胞的颗粒细胞都具有寡克隆起源,成熟卵泡中的颗粒细胞群是由最初的包绕卵母细胞的3~5个具有颗粒细胞潜能的细胞发育而来。颗粒细胞是卵巢雌二醇、抑制素和激活素的主要来源,并为卵母细胞发育成熟提供必需成分。但由于卵泡基底层将颗粒细胞与卵泡膜的血管分离,形成一个相对的血液屏障,故颗粒细胞不直接接受血液供应,所以限制了白细胞和高分子物质(如低密度脂蛋白)的进入。因此,相邻的颗粒细胞和卵泡细胞间的细胞连接作用尤为重要。
颗粒细胞通过广泛的缝隙连接网络相互连接,为相邻细胞之间的小分子代谢交换和传输提供重要途径。每个颗粒细胞间隙连接的数量随着卵泡的发育而增加,将它们连接成一个功能性整体。此外,颗粒细胞通过细胞质突触穿过透明带,与卵母细胞的细胞膜形成缝隙连接。缝隙连接由称为连接蛋白的六连环蛋白质组成。Connexin-37和Connexin-43是最重要的两种卵泡连接蛋白,它们形成具有不同通透性的间隙连接。
Connexin-37是卵母细胞中的主要连接蛋白,而Connexin-43在颗粒细胞中占优势,然而至少围绕卵母细胞周围的第一层颗粒细胞也表达Connexin-37。颗粒细胞与卵母细胞之间的通信是通过同型Connexin-37缝隙连接而进行的,而颗粒细胞之间的缝隙连接是通过同型Connexin-43复合物进行的。卵丘细胞局部诱导颗粒细胞Connexin-43表达。卵泡刺激素(follicle-stimulating hormone,FSH)TGF-β1和卵丘细胞分泌的全反式维甲酸均促进颗粒细胞Connexin-43表达。在窦卵泡中,颗粒细胞通过缝隙连接运输cGMP来抑制卵母细胞恢复减数分裂。LH的排卵高峰抑制Connexin-43 mRNA的表达,并导致丝裂原活化激酶介导的Connexin-43的磷酸化,最终导致缝隙连接关闭,细胞间代谢偶联中断。
Connexin37和Connexin43敲除的小鼠的卵巢表型进一步阐述了连接蛋白对卵泡功能的重要性。在通过靶向 Gja4 基因构建的Connexin 37缺乏型小鼠卵巢中,卵泡生长停滞在窦前卵泡;卵母细胞的生长虽然开始,却在减数分裂能力恢复之前停滞,导致卵母细胞的丢失和黄体化结构的形成。以 Gja1 基因为靶点构建的Connexin-43缺陷小鼠具有卵巢功能的异常,特征是生殖细胞数量减少和初级阶段以后卵泡生长受损。其他连接蛋白亦在卵巢中表达,但它们的特异性功能尚不清楚。
颗粒细胞表达大量细胞因子的受体并对这些因子做出反应,这些因子要么来源于卵泡局部,要么从血液进入卵泡腔。这些因素包括卵母细胞衍生因子、颗粒细胞自身产生的自分泌/旁分泌因子、卵泡膜细胞的产物以及来源于垂体和其他组织(如脂肪)的循环因子。除了FSH和LH,已证实体内、外存在众多影响灵长类动物以及其他动物颗粒细胞的信号分子,包括下丘脑因子[如促性腺激素释放激素和吻素(kisspeptin)]、其他垂体激素(如生长激素和催乳素)、大量生长因子[如表皮生长因子(epidermal growth factor,EGF)]家族成员、TGF-β家族成员、胰岛素样生长因子(insulin-like growth factor,IGF)和控制代谢的激素(如胰岛素)、血管张力素、血管生成因子、细胞因子(如TNF-α)和脂肪代谢相关因子(如瘦素、脂联素)等。
根据与人类突变或动物自发或诱发基因突变相关的表型研究中阐述了这些因子的关键作用,如前所述的GDF-9和BMP-15。但这些因子作用的主次和时间顺序还未得到很好的阐述。除了颗粒细胞表面受体的表达,微管系统和外泌体亦参与卵泡内的信号通信,例如以microRNAs为代表的外源性调节因子等。
此外,虽然前文提出颗粒细胞都是寡克隆起源,但根据其在卵泡内的位置而表现出显著的表型差异。主要由于它们与卵母细胞和卵泡膜细胞的距离不同,对其释放的旁分泌物质反应而导致位于基底层附近的壁颗粒细胞、位于窦腔的颗粒细胞和卵丘颗粒细胞各自具有独特的特征。窦卵泡壁颗粒细胞表现出最大的类固醇生成活性。此外,排卵前卵泡壁颗粒细胞LH受体水平最高,最靠近窦腔的颗粒细胞类固醇生成酶的表达较低,而中间区域的颗粒细胞比窦壁颗粒细胞具有更大的有丝分裂活性。
在排卵时随卵母细胞释放的卵丘细胞不表达芳香酶,其LH受体含量和LH反应性水平明显低于壁细胞。在小鼠中发现,卵丘细胞具有独特的基因表达模式,包括编码钠偶联中性氨基酸转运体基因 Slc38a3 的表达,以及更高水平AMH的表达。在卵母细胞分泌的GDF-9和BMP-15的作用下,靠近窦腔的卵丘细胞和颗粒细胞不表达mTOR信号传导的抑制剂DDIT4L,而壁颗粒细胞表达高水平的该蛋白。因此,卵丘细胞中的细胞代谢调节剂mTOR活性最高,使其更好地为卵母细胞的生长发育提供所需要的营养物质。卵丘细胞在LH峰后增殖,在排卵刺激产生的前列腺素的作用下,产生由透明质酸、蛋白聚糖和蛋白聚糖结合蛋白组成的细胞外基质。这种基质的形成导致卵丘-卵母细胞复合体的排卵前扩张,为排卵做准备。此外,不同颗粒细胞前列腺素E受体的不同表达模式允许颗粒细胞亚群在排卵期间对PGE 2 做出独特的反应。黄素化的卵泡颗粒细胞经过终末分化,产生大量的黄体细胞群。由于黄体新生血管化,颗粒-黄体细胞可从循环脂蛋白获取更多胆固醇,合成孕酮的能力显著增加。颗粒-黄体细胞还保留了从卵泡黄素细胞产生的雄激素前体合成雌激素的能力。
卵泡膜和间质细胞被认为是起源于卵巢间质中的成纤维细胞样间质细胞。它们首先出现在具有两层或更多层颗粒细胞的卵泡周围。GDF-9在卵泡膜的发育中起重要作用,在缺乏GDF-9的情况下,卵泡膜细胞可被募集到卵泡周围,但是无法进一步分化。Kit和Kit配体系统在卵巢和睾丸的雄激素产生细胞的早期发育中起着重要作用。卵泡膜细胞表达Kit配体的受体。因此研究人员推测颗粒细胞在卵泡膜细胞的形成过程中发挥重要作用。
此外,通过小鼠模型发现,hedgehog信号通路在卵泡膜细胞系的发育过程中也起着关键作用。在卵母细胞分泌的GDF-9的作用下,颗粒细胞分泌的DHH蛋白和IHH蛋白共同促进卵泡膜细胞的募集和分化。与FSH一样,角质形成细胞生长因子(keratinocyte growth factor,KGF)和肝细胞生长因子(hepatocyte growth factor,HGF)刺激颗粒细胞产生Kit配体,而Kit配体作用于卵泡膜细胞,促进KGF和HGF的正反馈表达。Kit配体、KGF和HGF在大窦卵泡中浓度最高。卵母细胞表达Kit受体——这种正反馈调节有利于更好地为卵母细胞生长发育提供养分。此外,卵泡膜细胞产生的胰岛素样因子3(insulin-like factor 3,INSL3)促进卵母细胞发育成熟。颗粒细胞产生的TGF-β家族成员在卵泡膜雄激素合成的局部调控中起关键作用。BMP-6抑制卵泡膜细胞基础的激素分泌和LH诱导的雄激素分泌。GDF-9(由颗粒细胞和卵母细胞表达)也抑制人卵泡膜雄激素的合成。排卵后卵泡膜细胞参与黄体的形成,称小黄体细胞,表达类固醇生成活性以产生雄激素前体。颗粒细胞则转化为较大的黄体细胞,表达芳香酶活性,进一步转化雄激素前体。
卵巢中的闭锁卵泡中有一些过度肥大的卵泡膜内层细胞。这些细胞被认为是去甲肾上腺素能神经调节卵巢类固醇生成活性的靶标。卵巢门间质细胞是具有较大结构的黄素样细胞,具有与睾丸间质细胞相似的结构的功能,像睾丸间质细胞一样,它们含有六角形的间质细胞晶体。门细胞与非髓鞘的交感神经纤维密切相关,这些细胞的内分泌活性被认为与女性青春期、怀孕期间以及围绝经期的类固醇激素分泌显著性相关。
卵巢基质含有表达某些类固醇生成酶的成纤维细胞。基质中也含有产雄激素细胞的前体。由于卵巢基质细胞表达雄激素受体,并可在雄激素刺激下增殖,因此卵巢来源高雄激素血症[例如,多囊卵巢综合征(polycystic ovarian syndrome,PCOS)和产雄激素的卵巢肿瘤将导致卵巢基质密度增加。基质细胞在卵巢中充当屏障作用,不仅在物理上将卵泡和黄体与相邻结构分离,还分泌大量生长因子和生长因子结合蛋白,发挥生物屏障作用,例如,卵巢基质细胞表达大量Gremlin,可以结合和灭活BMP;分泌卵泡抑素,结合和灭活激活素的卵泡抑素;分泌IGF-结合蛋白(IGF binding proteins,IGFBPs),和分泌结合WNT信号家族成员的卷曲相关蛋白等。
卵巢表面上皮是一种由中胚层细胞衍生来的扁平-立方上皮层,又称卵巢间皮,由于曾被误认为产生生殖细胞,被误称为“生发上皮”。成年卵巢表面上皮以黏蛋白基因 MUC1 的表达和表面的纤毛和顶端微绒毛为特征性表现。这些细胞位于覆盖致密结缔组织层的基底膜上。卵巢表面上皮在与腹腔的物质交换和排卵导致的表面缺损的修复中起作用。在排卵过程中,覆盖在卵泡上的上皮细胞经历凋亡,随后激活修复过程。这个过程包括在排卵后立即开始的细胞增殖以及细胞外基质成分的重新合成。促炎性细胞因子可能参与上述过程的调节。卵巢表面上皮内陷导致包涵体囊肿形成。这些包涵体囊肿中的卵巢表面上皮可发生化生,并可能发生瘤变。一些研究人员已经指出这些包涵体囊肿是由排卵部位周围内陷形成的,并且据报道,它们也与PCOS的发生密切相关。
卵巢生命周期的不同阶段都有巨噬细胞、淋巴细胞和多形核粒细胞的参与。这些细胞在维持正常的卵巢功能以及卵巢病理学发生方面发挥作用,例如,淋巴细胞浸润,尤其膜间质细胞的浸润,是自身免疫性卵巢功能障碍的主要表现。巨噬细胞是卵巢间质的主要细胞成分,通常出现在卵泡的毛细血管附近。在卵泡发育的早期阶段,在卵巢中很少观察到其他白细胞,但在排卵前期出现大量白细胞浸润,并与卵泡闭锁有关。肥大细胞在卵泡后期逐渐增多,释放组胺可能导致排卵期卵巢充血。排卵后,在趋化因子的作用下,嗜酸性粒细胞和T淋巴细胞募集迁移到黄体中。这些细胞的侵入和随后的激活发生在黄体退化之前。活化的T细胞产生吸引和激活巨噬细胞的趋化因子。在黄体细胞之间散布的暗星状K淋巴细胞曾被认为是巨噬细胞,它们通过直接的细胞-细胞接触和产生生长因子及细胞因子调节黄体细胞的功能。调节性T细胞,即维持抗原特异性T细胞耐受的淋巴细胞群,在黄体中具有特殊的重要地位。它们在中期黄体中大量存在,发挥抗炎和促稳态的功能。调节性T细胞的丢失与炎症反应的发生有关,促进黄体溶解。T淋巴细胞亚型和巨噬细胞的浸润在黄体溶解过程中发挥重要作用,而妊娠期这种侵入显著延迟。同时,卵巢中的中性粒细胞、嗜酸性粒细胞、淋巴细胞、单核细胞/巨噬细胞和肥大细胞产生一系列细胞因子,包括多个白细胞介素家族成员、TNF-α、IFN-γ、GM-CSF和MIP-1α,在卵泡形成和黄体功能发挥着重要作用。
卵巢受到内源性和外源性神经的双重调节。外源性神经主要由交感神经、感觉神经和极少的副交感神经组成,通过血管门周围丛进入卵巢。外源性神经主要参与调节卵巢血供和病理状态的疼痛,如卵巢子宫内膜异位症以及卵巢细胞的内分泌功能。此外,有研究发现,PCOS患者卵巢膜间质细胞间的神经支配显著增强,而肾上腺皮质能受体的过度激活与卵巢高雄激素症的发病有关。
人卵巢内源性神经包括儿茶酚胺能神经元,表达酪氨酸羟化酶(儿茶酚胺合成中的限速酶)。这些神经元大多表达神经营养素受体。神经营养素(neurotrophin,NT)是一类参与调节神经存活和分化的细胞因子,主要作用于TRK原癌基因家族的高亲和力受体和低亲和力p75神经生长因子(NGF)受体。一系列研究显示,NGF在早期卵泡的发育中至关重要,NGF或者NGF受体NTRK1缺乏的小鼠始基卵泡的形成显著减少,卵巢中未形成卵泡结构的卵母细胞数量增加。神经营养因子4(neurotrophin-4,NTN4)受体也被称为NT-5和脑源性神经营养因子(brain-derived neurotrophic factor,BDNF),其表达缺乏亦与始基卵泡形成的减少有关。基因敲除的小鼠的研究进一步发现,NTRK1和NTRK2受体的表达是始基卵泡的发生和早期卵泡发育的必需条件,并且NGF的作用部分通过诱导FSH受体在卵泡发育中的表达而介导。小鼠颗粒细胞在LH和NT-4/5(不是NT-3)作用下表达BDNF,卵母细胞表达BDNF受体TRK-B,从而促进小鼠卵母细胞成熟,包括排出第一极体,并促进体外早期胚胎发育。总体来说,现有的研究提示卵巢内神经营养因子系统可能对卵泡的早期发育和卵母细胞成熟有重要作用。神经营养素及其受体也在人胎儿卵巢中表达,TRK-B受体定位于生殖细胞基质中。从接受促排卵的妇女抽取的人卵泡液中可检测到p75、NGF受体、BDNF、NT-4/5和NT-3。除神经营养素及其受体外,包括P物质、血红素-1、截短受体NTK1R-Tr和NTK2R在内的速激肽系统也在人颗粒细胞中表达。
胚胎干细胞具有多能性,在体外可产生卵母细胞样结构,提示卵巢干细胞群在成年后可能产生生殖细胞。虽然仍存在争议,但已有研究显示可以通过用DDX4 N端抗体进行免疫标记流式分选分离得到小鼠和人卵巢中少量的卵巢生殖干细胞。但该研究中使用的DDX4抗体的特异性受到质疑,以及与它们反应的抗原仍待确定。用上述方法分离的细胞在培养中具有分裂潜能,并根据其形态和卵母细胞特异性标记(包括DDX4和LHX8)的表达形成卵母细胞样结构。这些来自小鼠卵巢的卵母细胞样细胞在体外和体内能够发育成能完成受精和胚胎发育的卵母细胞,从而产生存活的子代。由人卵巢皮质分离的人卵巢皮质干细胞具有相似的表型。用绿色荧光蛋白(GFP)标记的谱系标记物转染卵巢干细胞,注射后5~6个月注入成年小鼠卵巢,形成表达GFP的卵母细胞。部分GFP标记的卵母细胞可以成功受精,并在特定条件下发展到囊胚阶段。
将从成人卵巢皮质组织中分离并标记有GFP的卵母细胞注射到人皮质中,或与来自皮质的游离细胞重新聚集,并移植到免疫缺陷小鼠体内。移植后7~14天,移植物内可见到具有减数分裂标志的卵母细胞样细胞,周围有体细胞。虽然这些卵母细胞样细胞的特性尚不完整,但这些研究确实提供了卵巢中罕见的多能干细胞群体存在的证据。但这些细胞在数量上并不能达到生产卵泡以防止卵巢自然老化的作用。卵巢干细胞对正常女性或卵泡池大小不同的卵巢功能障碍女性(如原发性卵巢功能不全或多囊卵巢综合征)的卵泡数量的影响尚待确定。不同类型的干细胞也同样可以被诱导发育成生殖细胞前体。例如,如果提供适当的转录因子和/或生长因子,人胚胎干细胞和诱导的多能干细胞将在培养中分化成原始生殖细胞样细胞。该类细胞目前正被用于人类生殖细胞特异性分化和发育机制的基础研究。
总体而言,成年卵巢中多能干细胞数量非常少,如果将其移植到卵巢组织中,在体外处理后可产生功能性卵母细胞。在生理条件下,这些罕见的干细胞对卵泡池可能并无显著补充。但在体外可以将不同类型的人干细胞诱导发育成与原始生殖细胞非常相似的细胞,最终可能在临床应用上有较大的前景。
卵泡的生长开始于始基卵泡脱离静息状态,从人胚胎第4~6个月卵巢形成一直持续到卵巢功能的终末期。虽然一些研究人员已经提出,先形成的卵泡先发生排卵,但更多的研究提示静息卵泡过渡到生长阶段更可能是一个随机事件,与卵泡发育过程中形成的时序无关。
卵泡生长的开始以形态学的变化为特征,包括颗粒细胞形状从扁平变为立方形,颗粒细胞增殖,卵母细胞增大以及透明带的形成。扁平颗粒细胞向立方形的转化促使其功能的转变,包括某些功能mRNA(如卵泡抑素mRNA)的表达。颗粒细胞增殖与立方化进一步促进卵母细胞直径的增加。人类卵母细胞直径的第一次显著增加时发现最大卵泡截面上有15个颗粒细胞。卵母细胞的生长伴随着透明带的形成。随后,卵母细胞的生长和卵泡直径的增加呈正相关,直到卵母细胞形成平均直径达到80µm的次级卵泡为止。此时卵泡直径110~120µm,并具有大约600个颗粒细胞。卵泡增长至这一大小时,卵母细胞的生发泡达到平均26~27μm的最大直径。当卵泡继续扩张并压缩周围基质形成卵泡外膜,前卵泡膜细胞在颗粒细胞分泌信号的募集下向卵泡外的基质迁移,颗粒细胞分泌的信号包括Kit配体和IGF-1等,从卵母细胞分泌的信号包括FGF2、血小板衍生生长因子(platelet-derived growth factor,PDGF)、GDF-9和BMP-15也促进卵泡膜细胞迁移和增殖。随着次级卵泡的形成,颗粒细胞表达FSH、雌激素和雄激素受体,并通过缝隙连接紧密相连。卵泡膜的形成与卵泡的血液供应的发展相关,来自动脉的血供在邻近卵泡基底膜处形成环状毛细血管网络。同时,卵泡膜细胞表达LH受体并获得合成类固醇激素的能力。此后次级卵泡组成窦前卵泡池,FSH依赖的卵泡从窦前卵泡池中募集进入下一阶段,大部分卵泡通过闭锁而丢失。
次级卵泡生长的一个重要组成部分在于卵母细胞的分化和生长。生长中的卵母细胞代谢活跃,可以合成丰富的mRNA和蛋白质以支持早期胚胎着床前的生长和发育。周围颗粒细胞直接通过跨透明带突触与卵母细胞表面形成缝隙连接,进行营养素、生长因子和其他分子的双向转运,精密调节卵母细胞的生长。卵母细胞的生长过程中,通过透明带蛋白的积极合成、分泌、组装,逐渐形成成熟的透明带;部分细胞器的数量逐渐增加,特别是线粒体,在完全成熟的人卵母细胞中线粒体数量可达到大约有40万个;相反,部分细胞器在卵母细胞成熟过程中逐渐丢失,包括在卵原细胞中存在的中心粒。除了数量的变化,细胞器的分布随着卵母细胞发育成熟过程而发生变化,例如线粒体、内质网和高尔基复合体在紧邻生发泡区域逐渐聚集。卵母细胞在生长过程中逐渐获得减数分裂的能力,虽然具体机制尚不清楚,但只有在卵母细胞体积达到临界大小后才获得此能力。具有减数分裂能力的卵母细胞具有某些确切的属性,包括细胞周期蛋白CDK1、细胞周期蛋白B和CDC25水平的增加,这些蛋白的调控阈值很可能是细胞周期恢复的先决条件。此外,卵母细胞发育成熟的过程需要大量的能量,这些能量主要来自由颗粒细胞通过间隙连接转运的ATP和能量底物以及卵母细胞内丙酮酸的氧化磷酸化。缺乏丙酮酸脱氢酶的亚单位PDH1A的小鼠卵母细胞由于不能进行丙酮酸的氧化磷酸化,降低了ATP和烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)的水平。这些卵母细胞可以生长并排卵,但不能成功地完成减数分裂。在老年妇女的卵巢中,类似的过程可能导致胚胎染色体的不分离、错误分离和非整倍体的形成。基因组印迹的重建开始于卵母细胞的生长期——直到植入前胚胎发育后才完成,由包括DNA甲基化在内的多种机制参与调控。在缺失KDM1B(组蛋白H3赖氨酸-4-脱甲基酶)的小鼠中发现,组蛋白甲基化也有助于母系印迹的建立。母本或父本等位基因的异常表达会导致卵母细胞或雄性生殖细胞中印迹重建过程的失败。这些基因表达的改变与几种人类遗传性疾病有关,包括Beckwith-Wiedemann、Prader-Willi和Angelman综合征。卵母细胞中母系印迹的全部丧失会导致完全性葡萄胎的形成。
除了获得恢复减数分裂能力,生长过程中的卵母细胞逐渐获得胚胎的发育潜能,包括支持着床前的胚胎发育和发育成熟的能力,以及产生高度稳定的母体mRNA库。在卵母细胞生长完成时,母体mRNA库转录主动沉默,蛋白质翻译受到抑制,显著减慢直到卵母细胞成熟或受精。转录沉默需要卵母细胞与卵丘颗粒细胞通过缝隙连接进行特殊通信,并伴随大规模染色质结构的改变,这对于正在生长的卵母细胞获得恢复减数分裂和发育成熟的能力至关重要。在转录沉默后,排卵前卵母细胞中的母源蛋白和mRNA储存,除了用来支持减数分裂的恢复和受精后的第一次分裂,还包括卵母细胞质重塑精子DNA的能力和产生增强的钙振荡的能力。此外还有很多发育潜能有待进一步阐述。
卵巢内一系列细胞因子作为激活剂或抑制剂在调节卵泡生长的早期阶段起关键调节作用。卵泡生长和发育的激活因子包括LIF、碱性FGF和kit配体。卵泡颗粒细胞产生的kit配体和作用于卵母细胞和卵泡膜细胞上的kit受体,是卵泡生长和卵母细胞生长的启动所必需的。给新生小鼠注射抑制kit配体相互作用的kit抗体,可干扰卵泡发育的原始阶段。相反,注入重组kit配体,即扩散型和膜结合型,加速了新生大鼠卵巢从始基卵泡向初级卵泡的转变。而抗米勒管激素、激活素A和趋化因子SDF-1/CXCL12通过其受体CXCR4抑制早期卵泡生长。通过探究 Gdf9 敲除的小鼠和 Bmp15 基因纯合突变绵羊的卵巢表型发现卵母细胞来源的蛋白GDF-9和BMP-15对颗粒细胞增殖具有物种特异性。两种基因缺陷的情况下,颗粒细胞大约两次加倍后都停止增殖,卵母细胞继续增长。然而,大卵母细胞最终退化,并被单层颗粒细胞包围。
磷脂酰肌醇-3-激酶(PI3K)/AKT通路和哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路是目前发现的在始基卵泡的激活中起着关键作用的信号通路。当这两种通路被破坏时,都会导致小鼠卵泡生长的异常激活和始基卵泡的最终耗竭。PI3激酶催化3,4,5-磷酸肌醇磷酸盐(PIP3)磷酸化,激活蛋白激酶PDK1,而蛋白激酶PDK1又磷酸化激酶Akt,最后导致FOXO3的磷酸化。由颗粒细胞产生的Kit配体作用于卵母细胞上表达的Kit,通过一系列的信号传导最终导致FOXO3的磷酸化和失活(从细胞核排出),导致卵泡生长启动所需的基因表达被抑制。 Foxo3 基因无效突变的小鼠不能抑制始基卵泡的激活,导致出生后不久始基卵泡全部激活,表现为随后的始基卵泡耗竭和卵巢早衰。
PDK1和PTEN是目前发现调节FOXO3磷酸化的主要成分。卵母细胞特异性PDK1缺失导致始基卵泡耗尽从而继发不育。PTEN是PIP3磷酸酶,因此是PI3K的负调控因子。卵母细胞中PTEN的缺失,消除了对PDK1和AKT激活的抑制,也导致始基卵泡生长激活和早期卵泡耗竭。
mTOR是PI3K家族的成员,包含两个功能不同的mTOR复合体,通过下游靶点调控转录、翻译和代谢,在始基卵泡激活过程中发挥重要调控作用。此外,mTOR通路受到TSC1/TSC2复合物的负调控。研究显示,敲除的小鼠的卵母细胞中 Tsc1 和 Tsc2 ,虽然始基卵泡池仍能建立,但是始基卵泡被过早地激活,卵泡加速耗竭最终导致不育。而mTOR抑制剂雷帕霉素可抑制 Tsc1 敲除的小鼠的卵泡激活。此外,丝氨酸/苏氨酸激酶LKB1可通过抑制mTOR途径来抑制始基卵泡的激活。尽管以上研究证实mTOR信号通路在卵泡生长调控中的重要性,但其上游启动因子仍待进一步阐述,具体的信号传导途径尚未阐明。
Hippo信号通路在抑制生长、决定器官大小和组织稳态中起关键作用。Hippo信号通路的转导途径涉及一系列蛋白激酶,包括MST1和MST2,其通过磷酸化适配子蛋白SAV,并与之结合形成复合物,磷酸化LATS1和LATS2,后者与适配子蛋白MOB1A/B结合并磷酸化转录共激活子YAP1和TAZ。磷酸化的YAP1/TAZ保留在细胞质中。Hippo信号通路的失活导致YAP1和TAZ的低磷酸化,被转移到细胞核内,与TEAD1-4结合调节转录,导致细胞增殖和/或存活。该通路与AKT和mTOR通路在卵泡发育中具有相同的作用。
Hippo途径mRNA和蛋白质存在于卵巢中,随着卵泡从始基卵泡阶段进入窦状卵泡阶段,其表达量减少。在AKT激活的情况下,Hippo信号通路的失活可诱导卵泡向窦状卵泡发育。敲除Hippo通路组分编码基因的小鼠表现为生育缺陷和卵巢形态异常,包括卵巢囊肿的发生,全基因组关联分析研究发现编码YAP1的位点是PCOS候选基因之一,进一步验证Hippo通路在卵巢中的重要作用。
因此,有学者建议使用PTEN抑制剂、PI3K激活剂或mTOR激活剂对AKT、mTOR和Hippo通路进行干预以治疗卵巢早衰。而现有的研究已经验证,通过针对上述通路药物的干预,可以实现人类和小鼠卵巢中始基卵泡的体外激活,更有研究报道了对卵巢早衰妇女卵巢组织的体外激活并进行辅助生殖获得活产胎儿的案例。
除了上述信号通路外,根据小鼠的功能基因组研究,已明确了一些在始基卵泡向初级卵泡的转变过程中起作用的转录因子,包括NOBOX、SOHLH1和SOHLH2。 Nobox 、 Sohlh1 或 Sohlh2 基因突变的小鼠在始基卵泡过渡到初级卵泡的过程中有缺陷而不育。此外,细胞周期调节因子CDKN1B在始基卵母细胞的核内起作用,以抑制卵泡的早期激活。
此外,垂体切除的动物卵巢中仍有早期卵泡的发育,提示了卵泡生长的起始不需要FSH。在FSHβ亚基或FSH受体基因失活突变的人和小鼠中,卵泡可发育至次级卵泡和早期窦状卵泡阶段,但比FSH活性正常水平存在时更缓慢且频率显著降低。并且通过观察恒河猴胎儿垂体切除发现卵母细胞耗竭,说明了垂体衍生因子(不一定是FSH)在灵长类胎儿卵泡生长和生存中发挥重要作用。此外,对啮齿类动物卵巢的研究表明,窦前卵泡是促性腺激素的反应器官。对免疫缺陷和性腺功能低下的小鼠进行人类卵巢移植,发现FSH是超过两颗粒层阶段的卵泡生长所必需的。因此,没有促性腺激素的情况下卵泡可发育至窦前卵泡阶段,但是FSH可能促进卵泡的生长。
卵泡的发育成熟是卵巢功能建立的基础,从始基卵泡发育成熟为优势卵泡大约要经过一年的时间。在相当长的一段时期(大约300天)中,卵泡的成长不依赖促性腺激素,卵泡的发育过程由体细胞和生殖细胞之间的旁分泌和自分泌的局部因素调控。同一批募集的卵泡在该过程中持续闭锁而减少,直至青春期在促性腺激素作用下,通过激活相关基因转录、转录后机制(包括微RNA)和蛋白质翻译后修饰的信号途径等方式促进卵泡成熟。
(沈 薇)
1.Kim B, Kan R, Anguish L, et al.Potential role for MATER in cytoplasmic lattice formation in murine oocytes. PLoS One, 2010, 5: e12587.
2.Jansen RP, de Boer K.The bottleneck: mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol Cell Endocrinol, 1998, 145: 81-88.
3.Edson MA, Nagaraja AK, Matzuk MM.The mammalian ovary from genesis to revelation. Endocrine reviews, 2009, 30: 624-712.
4.Monne M, Han L, Jovine L.Tracking down the ZP domain: From the mammalian zona pellucida to the molluscan vitelline envelope. Semin Reprod Med, 2006, 24: 204-216.
5.Baibakov B, Gauthier L, Talbot P, et al.Sperm binding to the zona pellucida is not sufficient to induce acrosome exocytosis. Development, 2007, 134: 933-943.
6.Avella MA, Baibakov B, Dean J.A single domain of the ZP2 zona pellucida protein mediates gamete recognition in mice and humans. J Cell Biol, 2014, 205: 801-809.
7.Li L, Baibakov B, Dean J.A subcortical maternal complex essential for preimplantation mouse embryogenesis. Dev Cell, 2008, 15: 416-425.
8.Tong ZB, Gold L, Pfeifer KE, et al.Mater, a maternal effect gene required for early embryonic development in mice. Nat Genet, 2000, 26: 267-268.
9.Yurttas P, Vitale AM, Fitzhenry RJ, et al.Role for PADI6 and the cytoplasmic lattices in ribosomal storage in oocytes and translational control in the early mouse embryo. Development, 2008, 135: 2627-2636.
10.Fernandes R, Tsuda C, Perumalsamy AL, et al.NLRP5 mediates mitochondrial function in mouse oocytes and embryos. Biol Reprod, 2012, 86: 138, 1-10.
11.Peng H, Chang B, Lu C, et al.Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS One, 2012, 7: e30344.
12.Alazami AM, Awad SM, Coskun S, et al.TLE6 mutation causes the earliest known human embryonic lethality. Genome Biol, 2015, 16: 240.
13.Xu Y, Shi Y, Fu J, et al.Mutations in PADI6 Cause Female Infertility Characterized by Early Embryonic Arrest. Am J Hum Genet, 2016, 99: 744-752.
14.Zheng P, Dean J.Role of Filia, a maternal effect gene, in maintaining euploidy during cleavage-stage mouse embryogenesis. Proc Natl Acad Sci U S A, 2009, 106: 7473-7478.
15.Nakamura BN, Fielder TJ, Hoang YD, et al.Lack of maternal glutamate cysteine ligase modifier subunit (Gclm) decreases oocyte glutathione concentrations and disrupts preimplantation development in mice. Endocrinology, 2011, 152: 2806-2815.
16.Wu X, Viveiros MM, Eppig JJ, et al.Zygote arrest 1 (Zar1) is a novel maternal-effect gene critical for the oocyte-to-embryo transition. Nat Genet, 2003, 33: 187-191.
17.Eppig JJ, Wigglesworth K, Pendola FL.The mammalian oocyte orchestrates the rate of ovarian follicular development. Proc Natl Acad Sci USA, 2002, 99: 2890-2894.
18.Li Y, Li RQ, Ou SB, et al.Increased GDF9 and BMP15 mRNA levels in cumulus granulosa cells correlate with oocyte maturation, fertilization, and embryo quality in humans. Reprod Biol Endocrinol, 2014, 12: 81.
19.Persani L, Rossetti R, Di Pasquale E, et al.The fundamental role of bone morphogenetic protein 15 in ovarian function and its involvement in female fertility disorders. Hum Reprod Update, 2014, 20: 869-883.
20.Yamamoto N, Christenson LK, McAllister JM, et al.Growth differentiation factor-9 inhibits 3′5′-adenosine monophosphate-stimulated steroidogenesis in human granulosa and theca cells. J Clin Endocrinol Metab, 2002, 87: 2849-2856.
21.Liao WX, Moore RK, Otsuka F, et al.Effect of intracellular interactions on the processing and secretion of bone morphogenetic protein-15 (BMP-15) and growth and differentiation factor-9. Implication of the aberrant ovarian phenotype of BMP-15 mutant sheep. J Biol Chem, 2003, 278: 3713-3719.
22.Inagaki K, Shimasaki S.Impaired production of BMP-15 and GDF-9 mature proteins derived from proproteins WITH mutations in the proregion. Mol Cell Endocrinol, 2010, 328: 1-7.
23.Peng J, Li Q, Wigglesworth K, et al.Growth differentiation factor 9: bone morphogenetic protein 15 heterodimers are potent regulators of ovarian functions. Proc Natl Acad Sci U S A, 2013, 110: E776-785.
24.Guo J, Shi L, Gong X, et al.Oocyte-dependent activation of MTOR in cumulus cells controls the development and survival of cumulus-oocyte complexes. J Cell Sci, 2016, 129: 3091-3103.
25.Mottershead DG, Sugimura S, Al-Musawi SL, et al.Cumulin, an oocyte-secreted heterodimer of the transforming growth factor-beta family, is a potent activator of granulosa cells and improves oocyte quality?J Biol Chem, 2015, 290: 24007-24020.
26.Shuhaibar LC, Egbert JR, Norris RP, et al.Intercellular signaling via cyclic GMP diffusion through gap junctions restarts meiosis in mouse ovarian follicles. Proc Natl Acad Sci U S A, 2015, 112: 5527-5532.
27.Juneja SC, Barr KJ, Enders GC, et al.Defects in the germ line and gonads of mice lacking connexin43. Biol Reprod, 1999, 60: 1263-1270.
28.Simon AM, Goodenough DA, Li E, et al.Female infertility in mice lacking connexin 37. Nature,1997, 385: 525-529.
29.Di Pietro C.Exosome-mediated communication in the ovarian follicle. J Assist Reprod Genet, 2016, 33: 303-311.
30.Navakanitworakul R, Hung WT, Gunewardena S, et al.Characterization and small RNA content of extracellular vesicles in follicular fluid of developing bovine antral follicles. Sci Rep, 2016, 6: 25486.
31.Merkwitz C, Lochhead P, Tsikolia N, et al.Expression of KIT in the ovary, and the role of somatic precursor cells. Progress in Histochemistry and Cytochemistry, 2011, 46: 131-184.
32.Robertson SA.Regulatory T cells in the corpus luteum—new players in fertility control?Biol Reprod,2012, 86: 26.
33.Chao MV.The p75 neurotrophin receptor. J Neurobiol, 1994, 25: 1373-1385.
34.Kawamura K, Kawamura N, Mulders SM, et al.Ovarian brain-derived neurotrophic factor (BDNF) promotes the development of oocytes into preimplantation embryos. Proc Natl Acad Sci U S A, 2005, 102: 9206-9211.
35.Garcia-Ortega J, Pinto FM, Prados N, et al.Expression of tachykinins and tachykinin receptors and interaction with kisspeptin in human granulosa and cumulus cells. Biol Reprod, 2016, 94: 124.
36.White YA, Woods DC, Takai Y, et al.Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat Med, 2012, 18: 413-421.
37.Nilsson E, Parrott JA, Skinner MK.Basic fibroblast growth factor induces primordial follicle development and initiates folliculogenesis. Mol Cell Endocrinol, 2001, 175: 123-130.
38.Albertini DF, Combelles CM, Benecchi E, et al.Cellular basis for paracrine regulation of ovarian follicle development. Reproduction, 2001, 121: 647-653.
39.Kanatsu-Shinohara M, Schultz RM, Kopf GS.Acquisition of meiotic competence in mouse oocytes: absolute amounts of p34 (cdc2), cyclin B1, cdc25C, and wee1 in meiotically incompetent and competent oocytes. Biol Reprod, 2000, 63: 1610-1616.
40.Johnson MT, Freeman EA, Gardner DK, et al.Oxidative metabolism of pyruvate is required for meiotic maturation of murine oocytes in vivo. Biol Reprod, 2007, 77: 2-8.
41.Shi L, Suetake I, Kawakami T, et al.Xenopus eggs express an identical DNA methyltransferase, Dnmt1, to somatic cells. J Biochem, 2001, 130: 359-366.
42.El-Maarri O, Buiting K, Peery EG, et al.Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet, 2001, 27: 341-344.
43.Ciccone DN, Su H, Hevi S, et al.KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature, 2009, 461: 415-418.
44.Nicholls RD, Knepper JL.Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet, 2001, 2: 153-175.
45.Castrillon DH, Miao L, Kollipara R, et al.Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science, 2003, 301: 215-218.
46.Jiang ZZ, Hu MW, Ma XS, et al.LKB1 acts as a critical gatekeeper of ovarian primordial follicle pool. Oncotarget, 2016, 7: 5738-5753.
47.Oktay K, Newton H, Mullan J, et al.Development of human primordial follicles to antral stages in SCID/hpg mice stimulated with follicle stimulating hormone. Hum Reprod, 1998, 13: 1133-1138.
随着卵泡的发育成熟、成熟卵子的规律排出,卵巢功能趋于成熟。此过程中,卵泡的多种细胞成分紧密联系,通过一系列高度复杂的方式协调互作,调节卵巢分泌类固醇和蛋白质激素,并控制卵母细胞的发育成熟。卵泡来源的激素既作为局部的自分泌因子和旁分泌因子来调节促性腺激素对卵巢的作用,亦作为内分泌信号来反馈调节促性腺激素的分泌,从而促进卵泡发育并在卵泡发育成熟后诱发促黄体生成素的排卵前高峰,同时调节女性生殖道以利于受精、胚胎植入和早孕。卵母细胞是具有全能性和高度特异性的细胞,经历减数分裂和受精,形成新的胚胎个体。尤其特殊的是,卵母细胞的减数分裂过程可以停滞多年,只有在漫长的成熟过程完成时才会再次启动。卵巢的一生中只有大约400~500个卵泡或少于千分之一被募集的卵泡最终发育成熟并进行排卵,大部分卵泡在发育成熟过程中闭锁。随着女性生理年龄的增加,调节卵泡在卵巢生命周期中消耗速度的有利因素在逐渐减少,卵泡耗竭加速。
在FSH的支持下,卵泡发育进入窦状卵泡阶段。窦腔及其中的卵泡液不仅担任卵泡中营养交换和废物去除的载体,形成卵丘-卵母细胞复合物完成生长和成熟的独特环境,更在排卵时卵丘-卵母细胞复合物的释放过程发挥重要作用。窦腔的发育需要水的输入,这可能是由水通道蛋白7、8和9形成的水通道介导的。由于通过水通道蛋白进行水的净转移是需要渗透梯度的,因此颗粒细胞被认为能够主动地传输离子来产生这种梯度。另外,糖胺聚糖在窦腔中的水解可以提高卵泡的渗透压并支持水的渗入。在排卵前5~6天,由于颗粒细胞增殖和窦腔的积聚,卵泡迅速膨胀,并移至卵巢表面。排卵前卵泡的加速膨胀可引起月经中期盆腔疼痛。
“募集”一词用来描述卵泡离开静止卵泡池开始生长的过程。然而,一些作者也使用这个术语来描述一群窦卵泡参与进一步的生长。有人提出,第一种情况称为初始募集,后一种情况称为周期性募集。由于生长的卵泡容易闭锁,可能导致脱离生长轨迹,因此周期性募集虽然是必需的,但不能保证排卵。选择是指成熟卵泡群减少到适合于特定物种的排卵配额的数量的过程。这个过程需要对从属卵泡进行负选择,同时对优势的卵泡进行正选择。
尽管传统观点认为在月经周期中卵泡会形成一个单一的队列波,但是越来越精密的超声研究发现会出现多个队列波。有学者通过对正常排卵妇女的连续经阴道超声监测以及早期卵泡募集的组织学研究提出了“波动理论”,该理论认为在卵巢周期中会招募两个或更多个队列波的4~14个直径≥4~5mm的卵泡,并且在排卵间期的最终队列波中出现注定排卵的优势卵泡。
在早期卵泡期,优势卵泡和队列中其他健康成员之间没有明显的形态差异。然而,与队列中的其他成员相比,优势卵泡的大小和其颗粒细胞的高有丝分裂指数具有显著的辨识度。优势卵泡的标志包括优势卵泡的卵泡液中可检测到FSH和显著的高雌二醇水平,FSH是窦前卵泡向窦状卵泡过渡的必需条件,也是窦卵泡的生存因子,在没有对卵泡使用FSH增敏的局部因子的情况下,FSH水平的降低会诱发细胞凋亡。在新月经周期开始时,由于孕酮、雌二醇和抑制素A水平下降,黄体后期FSH水平升高,促使卵泡发育成熟为窦卵泡需要一定阈值FSH浓度来维持生长,并且在黄体晚期达到该阈值水平。值得注意的是,FSH仅增加10%~30%即可跨越阈值,这表明颗粒细胞对于循环中FSH水平具有高度灵敏的感知能力。在缺乏LH的情况下,FSH可以诱导卵泡生长至排卵前大小(至少17mm),虽然此时雌二醇浓度下降,但抑制素的产生可诱导并恢复颗粒细胞对FSH的正常反应。
FSH可能通过体细胞或卵母细胞产生的生长因子间接促进颗粒细胞的增殖。例如,在啮齿动物中,FSH刺激下产生的雌激素可促进颗粒细胞的增殖。优势卵泡的早期迹象是其颗粒细胞的增殖速度大于非优势卵泡。此外,FSH对颗粒细胞主要作用是诱导芳香化酶的表达。因此,缺乏FSH的激活,即使为颗粒细胞提供芳香化雄激素前体,也无法产生雌激素。FSH还诱导细胞色素P450还原酶和17β-羟基类固醇脱氢酶1(17β-hydroxysteroid dehydrogenase 1,17β-HSD1)的表达,细胞色素P450还原酶将电子转移至芳香酶,而17β-HSD1是将雌酮还原为雌二醇的关键酶。FSH诱导排卵前卵泡的颗粒细胞中LH受体的表达。LHR mRNA在直径3~10mm的窦卵泡中可检测到,在排卵前卵泡的颗粒细胞中达到最高水平。相反,随着卵泡直径增加,颗粒细胞中FSHR mRNA水平下降。因此,在卵泡成熟的晚期,LH可以协同FSH促进卵泡成熟。这种特性可使得优势卵泡在FSH水平下降时完成其成熟周期,也使优势卵泡准备对排卵前的LH峰做出反应。
人类FSHβ亚基和FSH受体失活的突变体表型以及小鼠中该类基因的靶向性缺失的研究进一步证实了FSH在卵泡发育中的重要性。FSH受体纯合突变的女性具有高促性腺激素性腺功能减退的特征,表现为第二性征缺乏或发育不良以及高FSH和LH水平。具有该类突变的人卵巢表型与FSH受体和FSHβ亚单位敲除的小鼠的卵巢表型相似。在缺乏功能性FSHβ亚基或FSH受体的情况下,卵巢较小,卵泡发育一般不超过窦前阶段。
“优势”是指注定要排卵卵泡的状态及其在调节排卵配额大小中的作用。计划排卵的卵泡在前一个周期的黄体消亡后5~7天逐渐占据优势地位被选择。卵巢静脉中雌二醇水平在月经周期的第5~7天显著变化可以佐证优势卵泡的出现。即使在双侧卵巢竞争性排卵不利于竞争性卵泡生长的情况下,优势卵泡使得双侧卵巢的环境均不利于竞争性卵泡的生长,而优势卵泡仍能继续生长。
通过对灵长类动物或人类卵巢中成熟卵泡或黄体移除或消融实验可初步阐述调控优势卵泡生长的时间顺序。在月经周期的第8~12天破坏灵长类动物卵巢中的优势卵泡会使下一个排卵前垂体分泌的促性腺激素峰出现的时间延迟。相反,黄体中期进行黄体切除术(月经周期第16~19天)则可导致促性腺激素峰的提前出现。在女性中,从优势卵泡或黄体的消融到下一个排卵的间隔一般是14天。这些结论与优势卵巢(即含有优势卵泡或黄体的卵巢)的周期结构是月经周期的计时器的概念相一致。因此,28天的月经周期是主要卵泡(卵泡期)和黄体(黄体期)固有寿命的结果,而不是由大脑或垂体决定的时间。
在月经周期的早期,预定排卵的卵泡就已经被选定了。卵泡群中没有其他成员有能力作为其替代物,此时优势卵泡被破坏将不能及时诱导月经中期的促性腺激素峰。在黄体期,下一轮卵泡的生长只有在黄体的干扰被去除后才会发生,无论是自然的(黄体溶解)还是人工的(黄体切除术)。通过灵长类动物的黄体切除术后激素替代研究表明,孕激素是抑制黄体期卵泡生长的主要因素,此外黄体分泌的抑制素A也能抑制FSH,从而抑制卵泡的发育成熟。卵泡发育过程的核心过程是血管的形成,抑制血管内皮生长因子(vascular endothelial growth factor,VEGF)可通过抑制卵泡血管形成或使血管通透性降低,限制卵泡生长所必需的关键生长因子或激素的获得从而抑制卵泡成熟。
优势卵泡具有显著的内分泌特征。直径<8mm的卵泡表现为相对低的卵泡内雌激素-雄激素比率,但是从中卵泡期开始,这个比率逐渐倒转。“选定的”优势卵泡能够合成足够数量的雌二醇进入血液循环,并且早在周期的第5~7天两侧卵巢雌激素分泌表现出显著不对称。雌二醇的局部浓度与卵泡大小直接相关,当循环中的雌二醇水平达到高峰时(大约1µg/ml),雄烯二酮的浓度相应降低。同时,孕激素和17α-羟孕酮浓度增加,导致早期颗粒细胞黄体化。抑制素A在卵泡液中的浓度随着卵泡成熟而增加,而抑制素B、激活素A和游离卵泡抑素则没有随卵泡大小而变化。因此,随着卵泡的成熟,从以激活素为主的环境转变为以抑制素A为主的环境。抑制素A水平的增加与颗粒细胞中抑制素Aα和β亚单位mRNA表达的增加有关。
IGF是低分子量、单链多肽生长因子家族的成员,因其与胰岛素的结构和功能相似而得名。IGF-1和IGF-2均存在于人卵泡液中。卵泡液IGF-1很可能主要来自血浆。而IGF-2则是由所有卵泡的卵泡膜和卵泡周血管以及小窦卵泡的颗粒细胞和卵泡膜细胞产生的,并且在排卵前的颗粒细胞中大量表达。研究显示缺乏IGF-1的小鼠,其卵泡成熟被阻滞,动物不孕,颗粒细胞增殖处于基础状态,对雌激素的反应降低。莱伦氏综合征女性以IGF-1缺乏为主要特征,给予GnRH类似物后可诱导卵泡成熟、卵母细胞发育,并可通过人类绝经期促性腺激素(human menopausal gonadotropin,hMG)诱发排卵。表明对于人类,IGF-1不是正常卵泡发育所必需的,IGF-2在一定程度上可作为其替补。即使给予比正常水平高2倍的IGF-1,恒河猴的卵巢功能也不会受到影响。因此,局部产生的IGF似乎足以维持正常的卵巢活动,并且升高的IGF水平不会破坏卵巢功能。IGF-1受体存在于优势卵泡的颗粒细胞中,IGF-1和IGF-2均能激活IGF-1受体。虽然IGF-2受体存在于卵泡膜和颗粒细胞中,但目前发现可能与信号传导无关。IGF-1和IGF-2可促进体外培养人颗粒细胞和颗粒-黄体细胞的DNA合成、细胞增殖和甾体激素生成。然而,这些体外实验是在限制性培养基中进行的,因此添加一般营养因子可以增加预期的细胞功能。IGFs的作用是通过结合蛋白的局部修饰来调节的。迄今为止已经发现的7个IGFBP中,至少有5个在人卵巢中表达。IGFBPs结合IGFs并中和它们的活性,它们也可能对卵巢细胞有直接作用。IGFBP-1、-2、-3、-4和-5是在卵泡液中直接检测到或通过分析颗粒细胞的mRNA而被鉴定。在这些结合蛋白中,IGFBP-4尤其令人感兴趣,因为它是FSH刺激人颗粒细胞产生雌二醇的有效拮抗剂。IGFBP-4也存在于闭锁卵泡中,提示该蛋白在卵泡闭锁过程中发挥作用。
促性腺激素和IGF抑制IGFBPs的分泌,从而增强IGF的生物利用度和促性腺激素作用。此外,IGFBPs的蛋白水解酶是控制IGF生物利用度的另一种机制。IGFBP-4蛋白酶的表达仅限于健康卵泡和黄体。妊娠相关血浆蛋白A(pregnancy associated plasma protein A,PAPP-A)是一种大分子糖蛋白,其金属蛋白酶活性可将IGFBP-4降解为非活性片段。小卵泡的颗粒细胞分泌低水平的PAPP-A,而优势卵泡的颗粒细胞分泌高水平的PAPP-A。缺乏PAPP-A的小鼠表现为较低的血清雌二醇水平、较低的血清孕酮水平和甾体激素合成酶表达降低有关,导致促排卵方案中获得的卵泡数减少。
通过对雌激素合成缺陷的病例的研究,不难发现雌激素在卵巢功能中的重要作用。虽然研究有限,但17α-羟化酶(cytochrome P450 17A 1 ,CYP17A1)缺乏的妇女,卵泡膜细胞不能产生雄激素来支持颗粒细胞雌二醇的合成。在雌激素缺乏的情况下,外源性促性腺激素可促进垂体去除后卵巢中卵泡生长到排卵前阶段。同样的,严重性腺功能减退的女性给予外源性的FSH也可以促进卵泡发育。虽然卵泡可以生长,但是在没有外源性LH情况下,卵泡中雌二醇合成是受限的。此外,类固醇生成急性调节蛋白(steroidogenic acute regulatory protein,StAR)、CYP17A1和芳香化酶缺陷的妇女也常表现为卵泡发育过程中的低雌激素水平。因此,虽然雌激素在卵泡的成熟过程中至关重要,但是对于卵泡发育至排卵前期的大小影响不大。
仅仅通过人雌激素合成相关酶缺陷疾病的研究无法确认从低雌激素分泌的卵泡中获得的卵母细胞是否具有能够发育成可以受精的卵母细胞并发育为胚胎的特性。但芳香化酶抑制剂的动物研究表明,雌激素对卵母细胞功能至关重要。在卵泡成熟期间对恒河猴使用大量芳香化酶抑制剂降低循环雌二醇水平对卵泡生长大小没有影响。然而,所得卵母细胞大部分只能恢复到MⅠ期前期,发育到MⅡ期的过程显著延迟。但是,这是雌二醇缺乏的直接反映,还是使用芳香酶抑制剂(1,4,6-雄三烯-3,17-二酮)的结果,还是由于雌二醇下降引起的内分泌状态的补偿性变化的结果尚不清楚。然而,竞争性芳香化酶抑制剂如来曲唑的临床经验表明,在卵泡生长过程中芳香化酶的抑制对卵母细胞的成熟没有主要的负面影响,从侧面说明了雌二醇对卵母细胞成熟的重要性。
虽然人类卵巢中多种细胞表达雌二醇的受体,但是雌激素在卵泡健康、成熟和黄体功能中的生理作用还有待阐明。卵泡中表达雌激素受体α、β1和β2,后两种受体类型在黄体化颗粒细胞表达更突出,但在排卵前卵泡腔中极高水平的雌二醇(约1μg/ml)引起了对经典雌激素受体系统的功能质疑——哪种受体可被配体完全饱和?此外,如上所述,卵泡生长本身并不需要高水平的雌二醇。
雄激素对灵长类动物的卵巢有多种作用。给予外源性的睾酮或5α-二氢睾酮可促进恒河猴卵巢中初级卵泡的生长和卵泡的存活,提示雄激素的促卵泡发育的作用。在这个模型中,雄激素受体在健康的窦前卵泡和窦卵泡的颗粒细胞中表达丰富,在卵泡膜和基质中表达较少。此外,雄激素受体表达与细胞增殖标志物Ki-67呈正相关,与细胞凋亡呈负相关。因此一些研究者建议将补充雄激素作为对促性腺激素反应不良的治疗方法之一。然而,这些益处不是脱氢表雄酮或其雄激素代谢物对卵巢直接作用的结果。甚至,一些体外研究中亦发现雄激素可抑制颗粒细胞增殖而促进卵泡闭锁。
雄激素对人卵泡功能有不利影响的证据包括卵泡液中高水平的5α-二氢睾酮和低雌二醇水平是闭锁卵泡的特征。有报道称卵泡内高浓度的5α-还原雄激素(如5α-二氢睾酮),可作为颗粒细胞芳香化酶活性的竞争性抑制剂。在这个理论基础上,多囊卵巢综合征患者的卵泡中5α-还原酶活性比正常卵巢的要高。因此,雄激素可能通过雄激素受体以及非受体介导的机制以阶段依赖性的方式对卵泡的生长和功能产生正负两方面的影响。
随着从优势卵泡中释放出的雌激素引发LH峰,及较小幅度的FSH升高。这触发了卵母细胞减数分裂恢复、排卵和黄体的形成。卵巢间质细胞、白细胞和巨噬细胞,通过释放基质降解酶和细胞因子,包括白细胞介素IL-1β和TNF-α在此过程中发挥重要作用。
在正常的月经周期,FSH诱导排卵前卵泡颗粒细胞上LH受体的表达允许LH在卵泡成熟的终末阶段接替FSH的主导作用。这些受体还使颗粒细胞能够对LH峰作出反应,LH峰启动了卵母细胞减数分裂的恢复、排卵和随后的颗粒细胞和卵泡膜细胞的黄素化。只有当LH水平达到阈值浓度时才触发这一系列生物学过程的发生。一定量的LH刺激卵泡膜雄激素产生,并与FSH协同作用促进卵泡成熟,高水平LH促进尚未达到Graafian阶段的卵泡提前黄素化,并进一步诱导卵泡闭锁,组成了卵泡成熟过程“LH窗口”的概念。这个概念为促排卵提供理论基础。刺激优势卵泡成熟的LH水平限制小卵泡的生长并抑制芳香化酶活性。因此,理论上可能通过使用LH或hCG来驱动终末阶段卵泡的成熟并限制多排卵的发生。
排卵-黄素化程序的启动是颗粒细胞和卵泡膜细胞根据细胞信号强度反应的结果(即cAMP增加的幅度),也可以通过激活补充cAMP的辅助信号级联通路来激活。LH受体激活cAMP和IP3信号传导具有剂量依赖性,是激活磷脂酶C所需的LH水平的10~100倍。这些信号通路影响非编码RNAs的表达,在协调排卵卵泡发育过程中的基因表达和对排卵期LH峰的反应中起着关键作用。
LH启动一系列排卵相关事件部分是通过丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路介导的。 Mapk1 和 Mapk3 双突变的小鼠表现为卵丘扩张、排卵、黄素化和减数分裂成熟缺陷而不育。 Lhr 敲除的小鼠卵泡发育停滞在早期窦卵泡阶段,发育的卵泡周围通常有一层相对正常的卵泡膜,但没有排卵或黄体化的迹象。该表型与LH受体基因( Lhcgr )纯合突变的女性相似,表现为正常的第一和第二性征,闭经伴循环FSH和LH升高。卵巢包含了从始基到窦卵泡的各阶段卵泡,卵泡膜细胞层发育良好,但没有排卵前卵泡或黄体。 LHCGR 的纯合子错义突变体(N400S)也被发现与空卵泡综合征有关。以上所描述的表型均表明,卵泡的正常分泌雌激素、卵泡的发育成熟和黄体的形成均需要LH,但卵泡膜细胞的形成不依赖LH。
此外,对人LH受体的激活突变体的探究进一步阐述了LH在卵巢功能中的作用。与男性的性早熟相比,具有这些突变的女性没有明显的生殖表型。曾有学者推测女性会出现高雄激素血症,类似于多囊卵巢综合征的表型。然而,研究发现卵泡各细胞成分发育均衡,卵泡膜细胞对LH受体并未过早发生活化反应,也未过早地出现黄素化,所以这类突变体的黄素化所需的cAMP或其他第二信使分子的水平可能与野生型不同。虽然LH在卵泡发育过程中的调控机制还有很多待探讨的细节,但LH峰在排卵中的作用是毋庸置疑的。
排卵前LH峰出现在卵泡破裂、排卵前最多38小时。在卵泡破裂之前,颗粒细胞和卵母细胞发生许多关键的变化,包括颗粒细胞增殖的抑制;间隙连接功能的丧失使颗粒细胞与卵母细胞间的电生理合胞体解耦联;颗粒细胞排卵所必需的基因的表达,包括编码EGF样因子两性调节蛋白、表调节蛋白和β-纤维蛋白等基因。在小鼠中,后一种生长因子激活EGF受体,诱导颗粒细胞中编码环氧合酶-2(cyclooxygenase 2,COX-2,也称为PTGS2)的基因表达,促进前列腺素雌二醇(prostaglandin E 2 ,PGE 2 )的合成。PGE 2 与EGF样因子协同作用,触发卵丘细胞形成富含透明质酸的基质,导致卵丘扩张。PTGS2和前列腺素受体EP2(prostaglandin E 2 receptor 2,PTGER2)缺陷的小鼠具有与卵丘扩张异常相关的排卵缺陷。
卵丘的扩张是排卵的关键过程,是由产生透明质酸骨架所需的基因、透明质酸合成酶2(hyaluronan synthase 2,Has2)、透明质酸结合蛋白聚糖变体(hyaluronan-binding proteoglycan versican,Cpg2)、TSG-6(Tnfaip 6)和穿透素3(Pentraxin 3,Ptx3)等共同介导的。支持卵丘扩张的基质是与TSG-6结合的透明质酸链形成的网状网络,可将血清衍生的α-胰蛋白酶间抑制剂复合物的重链转移至透明质酸,该复合物是由两个与硫酸软骨素和尿抑胰酶素共价结合的重链大分子。穿透素3是组织透明质酸基质的另一种成分——一种五聚体蛋白质。TSG-6和尿抑胰酶素缺乏的小鼠的排卵缺陷与卵丘扩张失败有关。
最终卵丘形成圆柱形的柱状结构上升并突出至卵泡表面,为卵泡的破裂作准备。卵泡的破裂伴随着卵子和卵泡液温和地(而非爆炸性地)排出,这表明卵泡液并没有处于高压之下。在灵长类动物中,由于黄体产生的孕激素对卵泡动力学的局部作用,排卵在卵巢之间交替进行。然而,这一理论并没有确凿的证据支持。虽然一些研究表明排卵在左右卵巢中的频率相等,但另一些研究表明右侧排卵更为频繁。总体而言,排卵的发生需要以下决定因素:
LH在排卵过程中的早期作用是在排卵高峰数小时内诱导颗粒细胞中孕酮受体(progesterone receptor,PR)的表达和孕酮的合成;核转录因子NR5A2(也称为LRH1)也部分促进了孕酮的合成。敲除小鼠的 Nr5a2 基因导致其卵泡不能排卵和黄素化。hCG通过蛋白激酶A途径上调体外培养的人颗粒细胞中孕酮受体的表达。在实验动物和恒河猴的研究中,通过合成孕酮受体拮抗剂和抑制孕酮的药物抑制动物排卵,提示了孕酮受体上调在排卵过程中的重要性。此外,在排卵前立即给予孕激素受体拮抗剂乌司他丁可以延缓妇女的卵泡破裂、排卵。靶向敲除小鼠的孕激素受体(尤其是A型受体,PR-A)以及缺乏功能性 Pparg 基因(受孕激素受体调控的基因)时,小鼠表现出排卵缺陷。此外,核受体相互作用蛋白RIP140(NRIP1)在小鼠的排卵过程中也发挥重要作用。NRIP1,最初被描述为转录抑制因子,其突变小鼠的卵丘细胞功能下降,而且控制卵丘-卵母细胞复合物扩展的基因表达方面存在缺陷。以上研究表明孕酮通过经典途径调节排卵相关基因的表达。然而,PR-A通常是转录的抑制剂,说明孕酮在排卵前期的重要作用也包括抑制基因的表达。已经鉴定出许多可能依赖于排卵期孕酮的候选基因,包括基质金属蛋白酶(matrix metalloproteinase,MMP)和组织蛋白酶L(cathepsin L)。PTGS2在孕激素受体缺陷小鼠中的表达是正常的,表明前列腺素不是孕激素调节排卵程序的一部分。
LH峰诱导排卵前颗粒细胞中PTGS2酶的表达,合成卵泡中的前列腺素。药理研究和基因靶向敲除小鼠的实验揭示了排卵过程中前列腺素所发挥的重要作用,全身或局部向窦状卵泡注射前列腺素合成抑制剂可抑制实验动物和恒河猴的排卵,导致未破裂的卵泡黄体化。而人体药理学实验进一步证实,与安慰剂治疗组在36小时内有明显的破裂迹象相比,口服选择性PTGS2抑制剂罗非昔布可将卵泡破裂的超声波征象时间延迟到LH高峰后48小时以上,进一步证实PTGS2在排卵过程中的重要作用。此外,由于可以通过给予外源性PGE 2 来对抗 PTGS2 基因靶向突变小鼠的排卵缺陷,PGE 2 被认为是参与排卵的关键前列腺素。 PTGS2 基因敲除的小鼠的异常表现之一是卵丘的扩张失败。针对PGE2的靶向受体EP2缺乏的模型小鼠也有排卵缺陷和排卵后卵丘扩张异常等表现。这些观察结果提示PGE2在排卵和卵丘扩张过程中发挥重要作用。孕酮受体在PTGS2缺陷小鼠中诱导了卵泡的形成,进一步证实了孕酮和前列腺素在排卵中的作用不尽相同。
LH可诱导EGF样生长因子双调蛋白、上皮调节蛋白、β-细胞蛋白的时序表达,利用上述生长因子体外培养啮齿类动物卵泡,可以重现LH引发的形态学和生化事件,包括卵丘扩张和卵母细胞成熟的必要过程。具有 Are (双调蛋白基因)或 Ereg (上皮调节蛋白基因)基因突变的小鼠在外源性促性腺激素作用下卵丘扩张显著减少。因此,EGF样生长因子是导致对LH促进排卵的卵泡反应的重要介质。
对于卵泡破裂的过程目前存在多种假说。有研究认为静水压的改变不是卵泡破裂的原因,因为通过直接测量静水压发现排卵前卵泡内压处于低水平,而胶体渗透压在排卵前卵泡显著增加,可能是由于颗粒细胞来源的蛋白聚糖浓度的变化。然而,窦状卵泡液成分的改变与卵泡扩大和破裂之间的因果关系尚有待确立。卵泡突触柱头的形成和破裂也提示了局部存在作用于卵泡壁的酶的作用,纤溶酶原激活剂和MMPs家族的成员是目前发现的排卵相关蛋白酶,将蛋白酶抑制剂滴入窦腔中可抑制排卵。排卵前纤溶酶原激活剂在大鼠卵巢卵泡壁中显著增加,提示其在排卵过程中的作用。然而,通过对缺乏尿激酶、组织纤溶酶原激活剂和纤溶酶原的小鼠的研究表明,纤溶酶对于卵泡破裂或者对于可能参与排卵的其他蛋白酶的激活不是必需的。
仅敲除 Mmp - 3 、 Mmp - 7 、 Mmp - 9 或 Mmp - 11 的小鼠可正常繁殖,表明这些酶单独在排卵中不具有强制性作用。然而MMP家族的其他成员(例如,MT1-MMP、ADAM-17)在卵巢中的作用尚不能从现有的敲除小鼠中确定,因为该类突变动物具胚胎致死性。
解离素、金属蛋白酶和血小板反应蛋白基序(a disintegrin and metalloproteinase and thrombospondin motifs,ADAMTS)家族的成员在排卵中亦起着关键作用。ADAMTS1在排卵前卵泡的颗粒细胞中表达,但在孕激素受体敲除的小鼠不表达,说明该基因是参与排卵的孕酮调节因子。由于卵丘扩张失败或活性生长因子的释放异常,小鼠 Adamts1 基因的靶向性缺失导致卵泡生长、排卵缺陷,从而导致雌性不育,其中ADAMTS4可能参与相关过程。 Cathepsin L 是另一种孕激素调节基因,可降解Ⅰ型和Ⅳ型胶原、弹性蛋白和纤维连接蛋白以及卵泡壁的所有成分,从而在排卵中发挥作用。
在给予排卵剂量的hCG 12小时后,恒河猴优势卵泡中MMP1、MMP10和MMP19、ADAMTS4、ADAMTS9、ADAMS15、Cathepsin L和尿激酶型纤溶酶原激活物等mRNA水平上调。此外,在给予hCG时向排卵前卵泡注射金属蛋白酶抑制剂(GM6001)可阻止排卵柱头的形成。上述研究结果表明灵长类动物的排卵也依赖于金属蛋白酶的活性。
卵母细胞的发育成熟伴随着排卵过程的发生,卵母细胞的成熟包括减数分裂的恢复和细胞质的成熟。从卵泡形成以来,卵泡可产生卵母细胞成熟抑制剂维持减数分裂停滞状态,从卵泡内环境中移出的卵母细胞会在培养过程中自发恢复减数分裂。而减数分裂抑制需要周围颗粒细胞的介导。尽管卵母细胞成熟抑制剂的全部生化性质仍然是个谜,但在小鼠模型中的实验已经发现许多下游介质负责控制减数分裂阻滞。与体细胞一样,卵母细胞的细胞周期是由现在称为周期蛋白和周期蛋白依赖性激酶的蛋白质水平和活性的变化控制的。在生物测定中,促成熟因子(maturation promoting factor,MPF)被定义为诱导减数分裂恢复的活性蛋白之一。将MPF通过显微注射入卵母细胞可导致减数分裂的恢复。进一步研究发现MPF是两种蛋白质的异二聚体:cyclin B和CDK1。MPF存在于发育成熟的卵母细胞中,但在排卵期LH高峰之前,其活性受到cAMP和cGMP的抑制。
卵丘细胞通过跨膜鸟苷酰环化酶NPR2与来自壁颗粒细胞的配体C型钠尿肽结合而产生cGMP的,cGMP通过间隙连接从卵丘细胞持续进入卵母细胞,一旦进入卵母细胞,cGMP抑制卵母细胞cAMP磷酸二酯酶3A(PDE3A),防止卵母细胞来源的cAMP水平的下降。卵母细胞中大多数cAMP是由卵母细胞质膜上的活性受体GPR3产生的。GPR3与刺激性G蛋白Gs偶联,激活腺苷酸环化酶,导致cAMP的持续产生。 Gpr3 敲除的小鼠的卵母细胞不能产生cAMP,因此其发生不依赖于LH的减数分裂恢复。卵母细胞中稳定的cAMP水平是细胞膜上腺苷酸环化酶产生的cAMP和细胞质PDE3A降解活性cAMP平衡的结果。卵母细胞来源的cAMP通过刺激cAMP依赖性蛋白激酶(cAMP-dependent protein kinase,PKA)的活性维持减数分裂阻滞。PKA至少磷酸化三种不同的蛋白质WEE1B、MYT1和CDC25B,从而抑制MPF活性并防止减数分裂恢复。颗粒细胞来源的cGMP阻断卵母细胞PDE3A活性,以及Gs复合体产生的cAMP激活PKA以维持MPF处于非活性状态,卵母细胞的减数分裂则不能恢复。
月经中期的LH峰在成熟窦卵泡中启动一系列事件,导致MPF的激活和减数分裂细胞周期的恢复。当生发泡由于核层的破坏而破裂时,卵母细胞核逐渐成熟并逐渐进入减数分裂Ⅰ期,随着细胞质的暴露,染色质浓缩,微管组织中心凝聚,然后聚集在纺锤体的两极。由肌动蛋白介导的细胞质流牵引纺锤体向皮层运动,随着含有卵母细胞一半染色体的第一极体的排出,第一次减数分裂完成。减数分裂细胞周期立即进入减数分裂Ⅱ期,然后在此阶段被阻滞;卵母细胞现在被称为次级卵母细胞或中期Ⅱ-阻滞卵(MⅡ期)。在排卵过程中,直到卵子从卵泡物理释放之前,减数分裂停滞在MⅡ期,直到受精第二次减数分裂才会恢复,并排出第二极体。
调节卵泡细胞和卵母细胞内cGMP和cAMP水平的细胞因子正是调节减数分裂的关键因素。月经中期LH峰介导的Gs耦合LH受体刺激卵泡细胞的Gs活化,并通过卵泡细胞跨膜腺苷酸环化产生cAMP。此cAMP随后激活转录因子,如cAMP-应答元件结合蛋白1(cAMP response element binding protein 1,CREB1)和cAMP-应答元件调节剂(cAMP response element modulator,CREM),这些转录因子诱导或抑制在卵母细胞成熟和排卵期间调节卵泡细胞功能的特定基因的转录。LH诱导的Gs活化还引起颗粒细胞鸟苷酰环化酶NPR2的脱磷酸化,从而降低其酶活性。颗粒细胞中cGMP水平的急剧降低使得cGMP能够快速地通过间隙连接从卵母细胞扩散出去,导致卵母细胞cGMP急剧下降。更低的卵母细胞cGMP水平不再足以使PDE3A失活,因此这种磷酸二酯酶变得活跃,破坏cAMP,导致抑制MPF诱导减数分裂成熟所需的PKA活性的损失。LH高峰之后随即发生卵母细胞和卵丘细胞之间缝隙连接的闭合。LH诱导的缝隙连接闭合部分通过壁层颗粒细胞合成和释放的表皮生长因子受体(EGFR)的配体双调蛋白及上皮调节蛋白介导,可促进间隙连接闭合所必需的EGFR激酶活性增加。卵泡细胞也分泌其他促卵母细胞成熟的旁分泌因子。例如,卵泡膜细胞分泌胰岛素样生长因子3,它可激活在卵母细胞质膜上表达的抑制性G蛋白(Gi)偶联受体RXFP2(也称为LGR8),减少卵母细胞cAMP的产生。
在低等生物中有明显的证据表明类固醇激素是诱导卵母细胞成熟的原因。但在哺乳动物中类固醇可能参与诱导减数分裂成熟,但抑制卵泡类固醇的产生或作用并不能阻止LH诱导的减数分裂的恢复,因此类固醇可能不是减数分裂恢复这一过程的必需物质。
LH峰除了触发卵母细胞MPF活性的改变外,还诱导卵母细胞中储存的母体mRNA的翻译。卵泡发育过程中这些母体mRNA与翻译装置一直处于隔离状态,直到减数分裂恢复才开始翻译表达。丝氨酸-苏氨酸激酶(serine/threonine kinase,STK)是这些新翻译的mRNA编码的蛋白之一,是一类细胞抑制因子的生物活性组成部分,因其通过显微注射到活跃分裂的细胞时会诱导卵母细胞中期阻滞而命名。MOS间接激活MAPK,在减数分裂MⅡ中抑制卵母细胞减数分裂发挥部分作用。MOS缺乏的小鼠生育力显著降低,卵母细胞在减数分裂MⅡ不能阻滞,在没有受精的情况下,小鼠卵母细胞继续分裂,从而形成畸胎瘤。随着充足的MOS产生,卵母细胞停滞在第二次减数分裂中期直至受精。受精时的钙振荡,引起细胞周期蛋白破坏和MOS蛋白降解,从而导致第二次减数分裂的恢复和第二极体的排出。
此外,现已发现在从甾脂醇到胆固醇的生物合成途径中,C29 4,4-二甲基甾醇中间体家族可诱导卵母细胞恢复减数分裂状态。从人卵泡液中提取的一种甾醇4,4-二甲基-5α-胆固醇-8,14,24-三烯-3β-醇被发现是减数分裂激活物质(follicular fluid meiosis-activating substance,FF-MAS)。从牛睾丸中分离到的4,4-二甲基-5α-胆固醇-8,24-二烯-3β-醇相关化合物,被命名为T-MAS。这些化合物是由 Cyp51 基因编码的P450 14α-脱甲基酶合成催化甾脂醇形成的。在排卵前卵泡液中,FF-MAS和T-MAS以微摩尔浓度存在:FF-MAS为1.6μm,T-MAS约为其的1/2。
FF-MAS和T-MAS在成熟卵泡中的累积可能与其合成增加以及在后续步骤中抑制胆固醇合成有关。据报道,促性腺激素可导致啮齿动物卵巢中 Cyp51 基因表达增加数倍,促进MAS形成。另外,排卵前期卵泡液中较高浓度的孕酮会阻碍卵泡发育后期胆固醇的合成,从而导致FF-MAS和T-MAS的累积。将FF-MAS灌注到啮齿动物卵巢后,可诱导卵丘细胞成熟而卵母细胞缺失或卵母细胞不成熟。然而,使用多种甾醇合成抑制剂的实验(包括阻断14α-去甲基酶的药物和抑制MAS代谢酶的药物)产生了矛盾的结果。14α-脱甲基酶抑制剂阻断了啮齿动物在促性腺激素刺激下的减数分裂,而阻断MAS代谢的药物通常导致卵丘内卵母细胞的生发泡破裂。因此,FF-MAS和T-MAS在卵母细胞成熟过程中的生理作用和药理作用仍然是不确定的。部分体外卵母细胞成熟相关的研究表明,这些化合物可通过刺激卵母细胞向减数分裂MⅡ期进展或提高卵母细胞的存活率而发挥作用,但不影响卵母细胞成熟。
细胞质成熟也发生在LH高峰之后,与核成熟相比,其形态变化不明显,但对于卵子的激活和着床前胚胎发育至关重要。在超微结构水平上,随着内质网、线粒体和皮质颗粒向卵母细胞皮质运动,卵母细胞细胞器的分布在此过程发生巨大变化。细胞器的运动由微管和微丝介导,并且依赖于细胞质网格的存在。当线粒体移动到皮层时,它们聚集在纺锤体周围,但在极体释放过程中,它们向卵母细胞导向的纺锤体极移动,因此大部分仍留在卵母细胞中。高尔基复合体断裂,解释了成熟卵母细胞合成新蛋白质能力的广泛下降。
随着染色质向皮质区域的移动,卵母细胞变得高度不对称。随着皮质区域肌动蛋白增厚覆盖在MⅡ中期的纺锤体上,肌动蛋白骨架发生改变。与富含微绒毛的卵母细胞质膜的其他部分不同,质膜的这一区域没有微绒毛。这种微绒毛的缺失可能减少精子进入减数分裂MⅡ纺锤体区域的机会,潜在地干扰减数分裂的正常进展。质膜金属锌是在许多细胞蛋白质(如锌指转录因子和金属酶)中起结构和催化作用的基本元素。在减数分裂成熟期间,通过质膜锌转运体吸收锌发生广泛的锌积累,卵母细胞锌含量总体增加约50%。在减数分裂MⅠ阻滞时,生发泡完整的卵母细胞通过干扰锌结合蛋白FBXO43(以前称为EMI2)的功能,导致细胞中有效锌的减少引起减数分裂的恢复,并导致皮质重组的异常和细胞极性降低。介导卵母细胞中锌储存的机制尚未被阐明,但可能涉及在亚细胞器中的隔离机制。
在分子水平上,细胞质成熟伴随着特定休眠的母体mRNAs的募集,这些mRNAs被翻译成蛋白质。在啮齿动物中,这些新募集的mRNA包括MOS、组织纤溶酶原激活剂和肌醇1,4,5-三磷酸1型受体(inositol 1,4,5-trisphosphate receptor type 1,ITPR1)。如上所述,MOS转录本的mRNA的翻译对于阻断细胞周期进展到减数分裂MⅡ期至关重要。小鼠的研究表明,IP3R-I蛋白随卵母细胞成熟过程中的表达增加可提高卵子钙振荡的能力,在卵子的减数分裂激活过程中发挥重要作用。而组织纤溶酶原激活剂的作用尚待阐明。
母体mRNAs募集的分子机制是胞质的多聚腺苷酸化。mRNAs的3′端的非翻译区中的特异性核苷酸序列,被称为胞质多聚腺苷酸化元件,指导这些mRNAs结合多聚尾[poly-(A)]聚合酶和poly-(A)的添加。调节该过程的蛋白质包括细胞质多腺苷酸结合元件蛋白1(cytoplasmic polyadenylation element-binding protein 1,CPEB1)和DAZL。多聚腺苷酸化导致特定母体mRNAs与多聚体的结合,mRNAs的翻译,从而增加编码蛋白的水平。胞质蛋白的翻译后修饰也发生在卵母细胞成熟过程中。例如,微管在从减数分裂MⅠ过渡到MⅡ期间发生乙酰化。微管乙酰化对于细胞器的正确定位和运动是必不可少的,可能通过影响微管蛋白和细胞质网格之间的相互作用来实现。细胞质蛋白的磷酸化和去磷酸化,特别是那些参与调节细胞周期的蛋白,是细胞质成熟的必要条件。
卵泡闭锁可发生在卵泡发育的所有阶段,是卵泡自发地或对环境因素或药物做出反应的结果。自发性卵泡闭锁主要是在卵泡形成或成熟的关键时刻缺乏必需营养因子(如FSH、IGFs等)的反映。在人胚胎卵巢中,生殖细胞的清除主要由细胞凋亡执行。成年卵巢的静息卵泡中,卵母细胞和颗粒细胞都参与凋亡,而在生长的卵泡中,颗粒细胞首先凋亡导致卵泡闭锁。在闭锁的生长卵泡中,凋亡的颗粒细胞逐渐累积,而在卵母细胞或卵泡膜中凋亡不明显。
一系列针对相应靶点突变的小鼠卵泡表型表明了凋亡在控制卵泡动力学中的重要作用。缺乏酸性鞘磷脂酶(一种产生促凋亡信号分子的神经酰胺酶)的小鼠卵母细胞储备增大,并且能够抵抗化疗药物和辐射引起的卵母细胞耗竭。Fas-缺陷( lpr / lpr )小鼠导致对FAS配体有反应性的卵泡表现为次级卵泡数量增加,大窦卵泡数量减少,有缺陷的卵母细胞和颗粒细胞死亡。FAS在闭锁的始基卵泡和初级卵泡的卵母细胞以及闭锁性窦卵泡的颗粒细胞中的表达丰富,与FAS在卵巢组织中卵泡闭锁性作用相一致。在缺乏促凋亡蛋白BAX的小鼠中,由于出生后细胞凋亡减少和颗粒细胞凋亡缺陷,卵母细胞储备增加。相反,缺乏抗凋亡蛋白BCL2的小鼠卵母细胞储备减少,敲除 Bcl - w 的小鼠或 Bcl - x 亚等位基因小鼠的卵巢也有同样的表型,缺乏细胞凋亡效应酶——半胱氨酸-天冬氨酸蛋白酶-2(cystein-asparate protease,Caspase-2)、Caspase-9和Caspase-11的小鼠由于胎儿生殖细胞凋亡下降,卵母细胞储备增加。 Caspase - 12 缺陷小鼠对抗癌药物诱导的生殖细胞凋亡具有抵抗力, Caspase - 3 缺陷小鼠由于颗粒细胞凋亡缺陷而表现出闭锁异常。此外,TNF-α和TNF-α相关凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)与颗粒细胞凋亡卵泡闭锁有关。
激酶PI3K和AKT被认为是主要的抗凋亡因子,它们通过磷酸化FOXO1和FOXO3来发挥功能。当被磷酸化时,FOXO1和FOXO3被排出细胞核外。当去磷酸化时,这些因子激活促凋亡基因的转录,包括FAS配体和BCL2家族的促凋亡蛋白,最终导致Caspases(Caspase 8、9和3)的激活。
排卵后,破裂的卵泡重组形成黄体。黄体形成和黄素化的过程伴随着大量基因的表达改变,仅在颗粒细胞中就包含数百种基因。破裂卵泡重组的显著特征是建立丰富的血管网。与卵泡破裂相关的排卵腔出血伴随有来自周围基质的毛细血管和成纤维细胞的渗透和增殖。由此产生新生血管能够使较大分子量的血液中循环的分子到达颗粒和膜黄体细胞,并有效分泌产物到循环中,如为黄体酮产生提供胆固醇底物的低密度脂蛋白等。黄体新生血管发展与孕酮的产生相互促进。当黄体完全形成时,血管内皮细胞约占黄体细胞含量的50%。
黄体的血管化由血管生成因子介导,包括由LH调控的血管内皮生长因子A(vascular endothelial growth factor A,VEGFA)和成纤维细胞生长因子2(fibroblast growth factor 2,FGF2)。研究显示,在排卵刺激后6小时内,猴卵泡液中的VEGFA含量增加6倍,并且在排卵后的36小时呈持续增长。由于在此期间VEGFA mRNA没有显著变化,因此VEGFA蛋白水平的升高可能是转录后机制调控的结果。
研究发现,给予促性腺激素处理的大鼠的可溶性VEGF受体(soluble fms-related receptor tyrosine kinase 1,sFlt-1)几乎完全抑制黄体血管生成,证实VEGFA在黄体血管网络发育中的重要作用。Notch配体和δ样配体4抑制VEGF介导的血管萌发和分支,体内抑制这些配体可导致黄体血管生成和血管密度的增强。同理在LH高峰之前,VEGFA也可在卵泡发育中起重要作用,因为给予VEGFA中和性抗体或FLT-1截短体会干扰排卵前卵泡的发育。基于血管生成素和TIE-2受体表达的时空模式,内皮细胞上表达的血管生成素和TIE-2受体也有助于黄体血管网的发展和维持。而抑制因子,如 VEGF 基因的剪接变异体及其可溶性受体则在调节促血管生成因子中起作用。
雌激素代谢物对黄体中的血管生成具有双向调节作用。这些代谢物既有促血管生成的作用(16-酮雌二醇和4-羟基雌酮),又有抗血管生成(2-甲氧基雌酮和2-甲氧基雌二醇)的活性。这些代谢物在人类黄体不同期分布不同,黄体早期和中期促血管生成代谢物浓度较高,黄体后期抗血管生成代谢物浓度最高。
壁层颗粒细胞在LH峰的作用下经历显著的形态学改变,统称为黄体化。随着颗粒细胞增殖相关基因表达的改变,这些细胞的有丝分裂潜能丧失:细胞周期蛋白D2表达终止,而细胞周期抑制剂 Cdkn1a 和 Cdkn1b 增加。同时,参与孕激素合成的蛋白编码基因(包括 STARD1 和 3BHSD2 )表达显著增加。人黄体类固醇生成细胞在大小和功能上具有异质性,包括黄体化颗粒和膜细胞。通过免疫组织化学和纯化后类固醇生成活性的研究提示这两种细胞具有不同的功能。颗粒黄体细胞表现出较高的孕酮的基础产量,由于其表达芳香化酶,因此推断是黄体雌激素合成的位点。卵泡膜黄体细胞含有17α-羟化酶/17,20-裂合酶活性,产生由颗粒-黄体细胞芳香化的雄激素前体,可能是黄体17α-羟孕酮产生的主要部位。因此,黄体中存在用于雌激素合成的双细胞系统。黄体中的大黄体细胞产生松弛素,一种被认为在促进子宫内膜蜕膜化、抑制子宫肌收缩活性和母亲对妊娠的适应性方面起作用的激素。
1)促黄体生成素的角色:
除了诱导排卵和黄体生成外,LH在维持黄体功能方面具有重要作用。在各种实验环境下长期撤去LH的支持几乎总是导致黄体的退化。猴在黄体期通过被动免疫或注入GnRH拮抗剂来撤去LH,均导致孕酮和其他类固醇激素水平显著下降。对处于黄体期的妇女注射GnRH拮抗剂6小时导致外周血孕酮水平显著下降。在猴模型中,如果只是暂时性的LH抑制,LH水平恢复后黄体孕酮的产生可恢复。在人类黄体的中晚期,从孕激素的分泌模式与LH的脉冲式释放模式一致可以看出LH对孕酮分泌的重要调控作用。
值得注意的是,短期抑制LH,只要能及时恢复,黄体的内分泌功能可以恢复,黄体根据其记忆模式,可以按正常存活14天。这种内在的记忆表明,黄体生成的过程会触发一个预定的生命周期,在没有受孕的情况下,这个生命周期就会按照预定的周期结束。这个记忆模式的分子和细胞机制仍有待阐明。有假说提出这种时间记忆模式是一系列连续的事件的结果,包括类固醇调节白细胞和免疫细胞的侵入,最终通过精细的细胞因子抑制黄体功能。
人黄体细胞膜LH/hCG受体的水平在黄体期逐渐升高,然后下降,但即使在黄体晚期仍然可被检测出。这种受体显然在内源性LH高峰后立即充分结合,因为排卵后几天内注射10 000U hCG不会引起孕激素水平的显著增加。然而,在黄体中期和晚期,外源性hCG显著增加黄体类固醇激素的合成。LH和hCG受体mRNA的表达趋向于遵循与LH和hCG结合相同的模式,从黄体早期到中期转录丰度增加,月经时下降。然而,如果怀孕,LH和hCG受体mRNA表达保持不变。在恒河猴中,促黄体激素受体的mRNA表达在黄体后期可维持到月经后才会下降。
2)孕激素的促黄体功能:
人黄体每天产生25~50mg孕酮。黄体细胞似乎对孕激素也有反应,所以它在生殖中既具有内分泌作用,又具有自分泌作用。在恒河猴中,孕酮受体拮抗剂米非司酮和HRP2000抑制hCG诱导的人颗粒-黄体细胞孕酮和松弛素的分泌。此外,可以用孕激素R5020治疗GnRH拮抗剂给药后恒河猴黄体中STARD1表达的减少。因此,LH和hCG对STARD1表达的作用可能是间接的,部分归因于孕酮的作用。
孕激素受体A型和B型均存在于恒河猴和人类黄体中,其mRNA从黄体早期到黄体中期表达逐渐增加,然后随着黄体年龄的增长逐渐下降。随着黄体年龄的增长,孕激素受体A与受体B的比值逐渐降低。上述孕酮受体拮抗剂对黄体细胞类固醇生成的作用可能是这些核受体调节的转录改变的一种反映。
黄体在非受精周期中的功能寿命通常为(14±2)天。黄体在非受孕状态下会转化为无血管瘢痕,称为白体。黄体退变,称为黄体溶解,包括一系列功能变化(即内分泌变化,最显著的是孕酮生产下降)和结构改变(即凋亡、自噬和组织退化)。LH的撤退和LH受体的下降并不说明灵长类动物的黄体溶解。然而,由于继发于LH和hCG水平的降低导致的受体表达下降,使灵长类黄体对hCG反应减弱。这种在黄体晚期的信号传导效率的降低所导致孕酮生产的下降与STARD1基因蛋白和mRNA水平的表达下降有关。黄体溶解期 Stard1 基因表达下降先于其他类固醇生成酶的表达下降。在黄体后期给予大量的hCG可使STARD1 mRNA和蛋白质水平达到黄体中期水平,导致血浆孕酮水平的急剧增加。注射成倍剂量的LH或hCG可延长猴黄体的寿命。这些研究表明,人类功能性黄体溶解的一个重要特征是STARD1表达的下降。高水平的hCG可以防止这种下降,维持孕酮的生产能力。虽然通过mRNA芯片的方式发现一系列关于类固醇摄入和甾体激素合成的mRNA在黄体溶解过程中表达改变,但由于不同的诱导黄体溶解方法[如GnRH拮抗剂、前列腺素F2α(PGF2α)]和自然黄体溶解过程所导致的表达改变缺乏一致性,导致结果的不确切性。
黄体的结构退化通过凋亡和自噬实现。早期黄体没有DNA断裂的证据,而中晚期黄体显示DNA断裂;与黄体中期相比,退行黄体中细胞凋亡的比率增加。相反,早孕的黄体组织未发现凋亡DNA片段。
控制人类黄体细胞存活和凋亡的因素仍然是一个有争议的话题。BCL2是一种细胞生存因子,主要分布在颗粒-黄体细胞、卵泡-黄体细胞、内皮细胞和血管中。一些研究人员没有发现在正常黄体期或使用hCG后BCL2水平变化的证据;然而,另一些研究人员描述了黄体期晚期BCL2水平的显著下降。据报道,凋亡前蛋白BAX在整个黄体期保持不变,并且从黄体中期的低水平增加到黄体退化期的高水平,而在妊娠的黄体中未检测到。如本节所述,FAS和FAS配体的表达在黄体退行时增加。已有资料表明,细胞凋亡是人类黄体退化的重要特征,一些报道描述了细胞存活( BCL2 )基因和凋亡前基因( BAX 和 FAS )表达的相互变化。然而,形态学研究发现自噬在黄体退化过程中发挥重要作用。相应的,研究发现包括 LC3 α、 LC3 β、 Atg3 和 Atg7 在内的自噬相关基因在牛黄体晚期的表达显著升高,而自噬抑制剂mTOR的表达显著降低。溶酶体激活和组织蛋白酶相关基因在黄体晚期表达也有显著增加。总体来说,迄今为止发表的研究表明,在退化的黄体中同时存在自噬和凋亡的激活。
那么,究竟是什么触发灵长类黄体对LH的敏感性降低以及随后在非生育周期中的黄体退化?虽然在实验动物中广泛认为PGF2α是黄体溶解素,但其在调节灵长类黄体退行性方面的作用尚不明确。PGF2α还可抑制体外培养的人颗粒-黄体细胞 STARD1 基因的表达。在体内,输注PGF2α会暂时降低人类黄体期的孕酮水平,黄体内注射PGF2α会导致孕酮产生下降和组织退化。黄体晚期表达PGF2α受体,PGF2α含量高于黄体早期。此外,PGF2α抑制hCG诱导的孕酮分泌,且在黄体后期最为显著。与家畜子宫来源的PGF2α参与触发黄体退化研究结果不同,人类的子宫切除术对黄体寿命没有影响。因此,参与人黄体退化的前列腺素可能不是子宫起源的,而可能是黄体本身。在猴体内,雌激素促进卵巢黄体溶解,并引起卵巢血中PGF2α水平升高。据报道,雌激素的黄体溶解作用可被吲哚美辛阻断;然而,其他研究人员已经提出,雌二醇在灵长类动物中的黄体溶解作用是通过对促性腺激素分泌的影响来介导的。有人提出PGE可通过抑制黄体PGF2α的表达而起到支持黄体的作用。当PGE在黄体晚期表达下降时,PGF2α表达增加。总体来说,这些研究提出PGF2α可能通过抑制孕激素的产生而在人类黄体溶解的启动中发挥潜在的作用。然而,这可能不是灵长类动物黄体退化的唯一介质。FAS和FAS配体mRNA和蛋白的时空表达与动物和人的黄体溶解密切相关。Fas-Fas配体系统可触发凋亡细胞死亡。在黄体后期,FAS蛋白的表达增加直至当黄体结构转变为白体。
有充分证据表明TNF-α超家族的细胞因子和干扰素-γ在人类黄体溶解中起作用。TNF-α在体外抑制人黄体细胞的类固醇激素的生成。来源于巨噬细胞和白细胞或内皮细胞TNF-α在黄体晚期显著增加。来源于巨噬细胞和白细胞的TNF-α可能部分通过上调内皮细胞来源的单核细胞趋化蛋白-1(MCP-1)发挥促黄体溶解作用。白细胞产生的其他促炎细胞因子也有助于抑制类固醇的产生。干扰素-γ在体外抑制促性腺激素刺激的人黄体细胞产生孕酮,并诱导细胞死亡。它是另一种巨噬细胞和白细胞的产物,有助于黄体的功能和结构溶解。正如前面关于卵巢白细胞和淋巴细胞的讨论中所指出的,充足的调节性T淋巴细胞可能对维持黄体功能和抗炎症状态至关重要,而且这种淋巴细胞数量的减少被认为促进了一种炎症环境,即细胞因子抑制黄体细胞类固醇生成。
黄体的血管成分可能通过产生直接或间接参与黄体溶解机制的因子(包括TNF-α、内皮素-1和MCP-1)来促进黄体退化。黄体溶解物质对内皮细胞功能的作用包括对细胞存活的影响可影响黄体灌注。在家畜的研究中发现,内皮细胞也可能是PGF2α的靶细胞,但人类黄体溶解过程中的作用尚未充分描述。
白细胞侵入黄体产生的活性氧是黄体溶解的另一个潜在重要因素。H 2 O 2 可导致人和大鼠黄体细胞孕激素分泌迅速减少,促性腺激素反应降低。H 2 O 2 的作用似乎是由OH-介导的,它抑制蛋白质合成,耗尽ATP,并诱导DNA损伤。H 2 O 2 使LH受体与腺苷酸环化酶解偶联,线粒体利用胆固醇生成类固醇的能力受损。
在受孕的周期中,人滋养细胞衍生的hCG的出现将黄体从溶解中解救出来。在晚期妊娠的黄体中,hCG抑制凋亡,对自噬的作用较小,允许黄体结构的维持和 Stard1 基因的表达。hCG在排卵后8天可在外周血中检测到,其浓度逐渐升高,既刺激类固醇生成,又防止黄体的结构退化,而黄体是孕酮最初10周的主要来源。妊娠期由于黄素化颗粒和卵泡膜细胞肥大,结缔组织和非甾体生成细胞,特别是内皮细胞的积累,黄体在妊娠最初6周的体积增大一倍。最近的研究表明hCG刺激黄体11β-羟基类固醇脱氢酶1型表达,导致黄体内皮质醇的生成增加,而皮质醇可能通过黄体细胞糖皮质激素受体在受孕周期中促进黄体存活。
黄体在妊娠最初几周是必需的,如果在妊娠7周之前进行黄体切除术,则会导致流产。然而,尽管存在hCG,但其分泌功能在整个妊娠期并不维持在高水平。17α-羟孕酮是一种不是由胎盘产生的类固醇,最大程度反映黄体的功能,通过监测其水平变化证实了上述观点。17α-羟孕酮水平在妊娠6周时达到高峰,然后下降。类固醇生成活性的下降部分归因于黄体早期细胞肥大随后萎缩的事实。妊娠黄体功能和结构变化的生化变化尚未阐明。
妊娠黄体瘤是一种罕见的良性肿瘤,通常发生在两侧卵巢的黄体化细胞,与hCG的影响有关。多囊卵巢综合征患者更常见。这些肿瘤产生的中重度的雄激素,导致母体出现高雄激素血症和女性胎儿男性化的症状。并分泌孕激素和雌激素。这些肿瘤在怀孕后会自动消退。黄体过度反应,通常表现为妊娠晚期由膜黄体囊肿引起的卵巢增大,是由多胎妊娠、葡萄胎妊娠或绒毛膜癌引起的高水平hCG所致。大约30%的病例与母体高雄激素血症症状有关。
除了产生成熟的配子以供人类繁衍外,卵巢的另外一个重要功能就是分泌激素。卵巢分泌的激素和细胞因子不仅通过自分泌及旁分泌调节卵泡的发育成熟,更参与全身多脏器功能的调节和维护。卵巢分泌的激素包括甾体激素和蛋白质激素。
甾体激素属于脂类化合物,是由多种酶联合作用对胆固醇按次序修饰、去除侧链、烯键位置的调整和羟基的添加而形成。基本结构为环戊烷多氢菲核,因结构与胆固醇相似,也称类固醇激素。卵巢产生的甾体激素主要包括雌激素、孕激素和雄激素。甾体激素合成的基本过程为类固醇生成急性调节蛋白(steroidogenic acute regulatory protein,StAR)将细胞摄取的胆固醇由线粒体外膜转移至相对缺乏的内膜,再由细胞色素P450、血红素蛋白混合功能氧化酶、羟甾脱氢酶、还原酶等对胆固醇进行一系列复杂的化学修饰而形成。
催化甾体框架发生变化、催化侧链的裂解以及羟基化和芳香化等。在氧分子和等价的还原物的参与下完成催化过程。产生甾体激素的细胞色素P450基因家族的每个成员都用cyp表示,其后用其作用部位碳原子的位置数字表示唯一性。
通过三个催化循环完成胆固醇侧链的裂解:前两个循环分别在碳-22和碳-20位置引入羟基,第三个循环导致碳间侧链断裂。
分布于内质网,主要催化孕烯醇酮和孕酮碳-17的羟基化,并将孕烯醇酮转化为碳-19的类固醇。
分布于内质网酶,它通过3个NADPH分子和3个氧分子催化碳-19底物的3个连续的羟基化反应生成一个含酚A环的碳-18甾体分子。
参与醛固酮的合成,分布于线粒体,分别由 CYP11B1 和 CYP11B2 编码,两种酶编码的蛋白质仅在33个氨基酸残基中有差异。这两种酶均有11β-羟化酶和合成醛固酮的活性,但P450c11AS可在氧化还原底物的协助下催化碳-18结构产生醛固酮。 CYP11B2 只在肾上腺皮质表达,而 CYP11B 除了在肾上腺皮质表达外,还在睾丸间质细胞和卵泡膜细胞表达,参与11-酮睾酮的生成。
以嘧啶核苷酸作为辅助因子还原酮基或氧化羟基。除了在产生甾体激素的细胞中参与激素的合成外,还与还原酶、甾体激素磺基转移酶以及甾体激素硫酸脂酶一起,调节靶组织中生物活性的激素水平。也是细胞对内源性甾体激素以及甾体激素类药物反应的主要决定因子。
是定位于内质网和线粒体的膜结合酶,以烟酰胺腺嘌呤二核苷酸(NAD+)为辅因子,催化3β-羟基脱氢和Δ 5 烯键异构化产生Δ 4 酮结构。进而将孕烯醇酮转化为孕酮,3-、17α-羟基孕烯醇酮转化为17α-羟基孕酮,将DHEA转化为雄烯二酮等。
是一类具有特定生物合成和分解代谢作用的酶,在甾体激素的合成和代谢过程中发挥重要作用。目前发现由14个17β-HSD的亚型,分别以发现的时间先后命名为17β-HSD1-14。在靶组织通常氧化17β-羟基类固醇使其失活的代谢过程,至少涉及7~14个17β-HSD。除了17β-HSD 5属于醛酮还原酶(aldo-keto reductase,AKR),其余都是短链脱氢酶/还原酶家族的成员,它们具有不同的辅因子和底物特异性,包括非甾体分子、亚细胞位置和组织特异性表达模式。虽然被归为一个家族,但编码17β-HSDs的基因结构不同,核苷酸序列同源性较低。根据其功能又可以被分为催化NAD+依赖的氧化酶(2、4、6、8、9、10、11和14型)和催化NADPH相关的还原酶(1、3、5和7型)。
又被称为雌激素17β-HSD,它通过减少弱雌激素活性的雌二醇来产生更高效17β-雌二醇,完成雌激素生物合成的最后一步。这种酶是一种使用NADH或NADPH作为辅助因子的胞质蛋白。它对碳-18类固醇的亲和力是碳-19类固醇的100倍。还可以将16α-羟基酯醇转化为雌三醇,显示出适度的20α-HSD活性。HSD17B1在卵巢颗粒细胞和胎盘合体滋养层细胞中表达。HSD17B1在乳腺癌细胞中的表达水平也高于HSD17B2,后者将雌二醇转化为雌酮。在雌激素受体反应性阳性乳腺癌中,表达HSD17B1的患者生存率低于HSD17B1不表达的患者。由于HSD17B1的晶体结构分辨率已被确定为2.2Å,可根据其分子结构设计特异性的抑制剂,用于雌激素反应性阳性癌症的化疗,具有重要临床意义。
定位于内质网酶,使激素失活,优先将睾酮氧化为雄烯二酮,并以NAD + 为辅助因子将雌二醇氧化为雌酮。表达在肝脏、分泌性子宫内膜、胎儿毛细血管内皮细胞、乳腺组织和大血管内皮细胞中。在正常乳腺组织中,HSD17B2的表达高于HSD17B1。然而,HSD17B2也能将20α-羟基孕酮转化为孕酮。
是一种内质网酶,被称为雄激素17β-HSD,以NADPH为辅因子,催化雄烯二酮为睾酮,使睾丸间质细胞完成睾酮生物合成的最后一步。还可以将雌酮还原为雌二醇。HSD17B3在卵巢中不表达,需要利用卵巢利用另一种酶(可能是HSD17B5)来合成睾酮。此外,由于17β-HSD的作用底物非常广泛,该家族中的部分酶主要在与类固醇代谢无关的基本代谢途径中发挥作用,这些酶的缺陷导致代谢性疾病的发生。
NADPH将Δ 4 甾体激素生成A环为饱和的甾体激素。甾体激素生成过程中关键酶的异常与一系列病理表型有关。与人胚胎睾丸相比,胚胎卵巢通常被认为是甾体生成的静息状态,尽管可检测到胆固醇侧链的裂解活性和CYP17A1酶活性,但直到青春期甾体合成能力才变得明显。
甾体激素一般为小分子物质,以扩散的方式进入细胞,与细胞核内特异性受体结合而发挥生物作用。目前研究显示,某些甾体激素也存在膜受体的表达,如雌激素。不同组织对同一甾体激素的应答方式具有相似的作用机制:性激素通过弥散的方式穿过细胞膜进入细胞内,与受体蛋白结合形成激素-受体复合物,受体构型改变而活化,活化后的激素-受体复合物形成二聚体进入细胞核,与靶基因上游的特异性DNA位点即激素反应元件(hormone response element,HRE)结合,激活RNA多聚酶,转录基因中的传导信息,生成特异的mRNA进入胞质内,在核糖体内翻译,生成基因编码的蛋白质,经剪切加工,形成有生物效应的蛋白质而发挥生物学效应。雌激素、孕激素均遵循以上基本作用机制并发挥生物学作用。
女性体内甾体激素主要在肝内代谢,其代谢速率与性激素结合球蛋白(sex hormone binding globulin,SHBG)结合容量成反比;肝脏或其他器官对甾体激素的摄取受激素与血浆中甾体激素结合蛋白和白蛋白的亲和力所影响,甾体激素与SHBG和皮质类固醇结合球蛋白(corticosteroid-binding globulin,CBG)的结合降低其外周代谢。游离甾体激素和白蛋白的亲和力相对降低,硫酸基结合的甾体激素和白蛋白结合紧密,因此从血中清除非常缓慢。故血中甾体激素硫酸盐的浓度通常比它们相应的非结合形式高数倍。相反,甾体激素葡萄糖醛酯与白蛋白结合力弱而被很快清除。卵巢分泌的载体激素主要包括雌激素、孕激素和雄激素三大类。
1.雌激素 雌激素是一类由碳-18雌甾烷骨架组成的甾体激素,由卵巢内的卵泡主要是优势卵泡分泌,其分泌的雌二醇(E 2 )占育龄期女性体内雌二醇总生成量95%,其生成率为90~250μg/d;分泌的雌酮(E 1 ),生物效能为雌二醇的1/3,占育龄期女性体内E 1 总生成量的50%,其生成率为110~260μg/d。育龄期女性血中雌激素水平呈周期性变化。一般月经周期第l周甚少,排卵前1天达第一个高峰,排卵后有所下降,月经周期21天左右,形成第二个高峰,待黄体萎缩时其水平急速下降,至月经前期达最低水平。雌激素的合成以卵泡膜细胞分泌的雄烯二酮为底物,在FSH激活颗粒细胞产生的芳香化酶催化下转变而成,这就是“两细胞/两促性腺激素理论”。颗粒细胞黄素化后也能分泌E 1 和E 2 。绝经后女性体内雌激素的主要来源是从外周转换而来的E 1 。肾上腺皮质亦能分泌少量雌激素。
雌二醇的代谢产物为雌酮及其硫酸盐、雌三醇。前两者仍有一小部分可转化成雌二醇,雌三醇则是不可逆的代谢产物。主要经肾排出,有一部分经胆汁排入肠内可再吸收入肝,即肠肝循环。
雌激素作用广泛,对自中肾管衍生而来的组织均有促进发育的作用。其主要作用是促进和维持女性生殖器官的发育以及第二性征的出现和维持。雌激素受体除分布在生殖道和乳腺外,还存在于肝脏、骨骼、心血管和大脑等器官。因此其主要作用体现在:
(1)生殖系统:雌激素参与卵泡生长发育各环节的调节,促进颗粒细胞的增殖和分化,雌激素的反馈调节与卵泡优势化有关。优势卵泡分泌雌激素能力强,其卵泡液中雌激素水平高。一方面,雌激素能协同FSH促进卵泡内膜细胞和颗粒细胞合成LH受体,协同FSH对颗粒细胞的作用,提高卵泡对FSH的敏感性。另一方面,雌激素对垂体FSH的分泌具有负反馈抑制作用,使循环中的FSH水平下降。因此,卵泡早中期,随着卵泡的发育和雌激素分泌的增加,垂体分泌FSH减少。此时分泌雌激素能力强,对FSH敏感的卵泡被优势化,反之则闭锁。雌激素也有助于子宫内膜腺体和间质增殖、修复。长期无孕激素拮抗的雌激素作用可引起子宫内膜过度增生或子宫内膜癌。此外也促进子宫肌细胞的增生和肥大,使肌层增厚,增加子宫肌层的血液供应,促使和维持子宫发育,增加平滑肌对缩宫素的敏感性。使宫颈口松弛、扩张,宫颈腺体分泌增多,含有的水分、盐类及糖蛋白增加,性状变稀薄,富有弹性,易拉成丝状,以利于精子的存活和穿透;促进输卵管肌层的发育及收缩,在雌激素的作用下,输卵管内膜的上皮细胞分泌活动增加和纤毛的生长,输卵管蠕动增强有利于卵子的输送;促使阴道上皮细胞增生、角化、黏膜变厚,并能增加细胞内糖原储存量,在乳酸杆菌作用下使阴道呈酸性,不利细菌在阴道内繁殖;促使大小阴唇增大丰满,并使脂肪沉积和色素沉着。
(2)乳腺及第二性征:青春期雌激素可刺激垂体前叶合成与释放催乳素,刺激乳腺导管和结缔组织增生,促进乳腺生长发育,并与孕激素、催乳素和肾上腺皮质激素协同,促进乳腺的发育和增加乳头乳晕的着色。诱导女性第二性征的形成:女性体态的形成、脂肪分布(臀、股部、乳房),骨盆、骨骼的宽大,声调,毛发特别是阴毛、腋毛的生长和分布等。
(3)下丘脑、垂体:对丘脑下部和垂体产生反馈调节,包括抑制性的负反馈和促进性的正反馈作用,从而间接对卵巢功能产生调节作用。
(4)代谢系统:①糖代谢:雌激素能增强葡萄糖刺激胰岛素分泌的反应,使得血浆胰岛素水平增加,降低糖耐量。②脂代谢:雌激素促进血浆胆固醇的降解和排泄,从而降低血浆胆固醇和β-脂蛋白的水平,提高血中载脂蛋白AI增加血清磷脂和α-脂蛋白的含量,降血胆固醇浓度。③蛋白质:雌激素能增强蛋白质合成,特别是促进生殖器官的细胞增殖与分化增强转录过程,加速蛋白质合成,从而促进生长发育。机体外周组织可发生氮潴留,并影响合成代谢,常因为雌激素不足而出现负氮平衡。④水盐代谢:雌激素参与孕酮和醛固酮的竞争作用而引起水和钠的潴留。⑤钙、磷代谢:雌激素与甲状旁腺激素共同维持血中钙磷的平衡。
(5)骨骼:雌激素促进儿童期长骨生长,加速骨骺闭合。直接促进成骨细胞功能,抑制破骨细胞分化及功能,抑制骨吸收和骨转化。此外,还能通过促进1,25-(OH) 2 D 3 的合成而增加肠钙吸收,促进降钙素的合成,对抗甲状旁腺激素的作用,保持骨量。雌二醇抑制成骨细胞的凋亡。在围绝经期由于雌激素分泌缺少,骨基质形成不足,钙盐无法沉积就会发生骨质疏松;牙齿脱落的发生与骨质疏松异曲同工,曾有研究表明:雌激素缺乏的女性落齿率高,而补充雌激素者落齿率明显下降。
(6)心血管系统:雌激素扩张血管,改善血供,维持血管张力,保持血流稳定。可通过改善脂代谢,降低总胆固醇和低密度脂蛋白(low-density lipoprotein,LDL),增加高密度脂蛋白(high-density lipoprotein,HDL)的含量,其引起的血脂变化,抑制动脉壁粥样硬化斑块形成。此外,通过对绝经后雌激素缺乏状态的研究表明:雌激素可通过直接作用,影响血管内皮功能、雌激素代谢物作用,影响肾素-血管紧张素系统、拮抗氧化应激作用以及抑制血管重塑等实现降压作用,维持血管张力保持血流稳定,从而达到保护心血管系统的作用。
(7)中枢神经系统:在中枢神经系统中,雌激素不仅作用于大脑中与生殖相关的神经回路而影响生殖过程,而且还作用于与学习、记忆、情感以及运动协调功能相关的神经回路,影响学习和记忆。近年来的研究还发现它不仅在神经系统的发育过程中具有神经营养作用,而且对成年人和动物的神经系统的功能活动也具有作用,并具有重要神经保护作用。循环雌激素水平的变化与女性的神经系统退行性病变之间关系密切。雌激素与中枢神经系统的结构和功能活动密切相关。生理性雌激素可以促进新生儿大脑发育以及成人的神经元生长和递质功能、阻止神经元细胞萎缩、调节突触的可塑性,此外,也影响人类各种心理活动,包括认知、心境、性欲以及攻击行为。因此,有人称雌激素为“天然的精神保护因子”,它可调节情绪相关脑功能。在胎儿发育早期阶段,大脑是唯一产生雌激素的器官。神经系统发育过程中,大部分脑区的神经元都表达雌激素合成过程中的关键芳香酶(主要集中在下丘脑的视前区、皮质、海马、中脑及杏仁体)。这些神经元内的芳香酶可以催化雌激素合成。发育过程中,雌激素不仅影响皮层神经细胞的发育,而且对海马的发育以及神经干细胞的增殖、分化也有重要作用。在生育期,中枢神经系统的雌激素受体参与多种活动,包括内分泌和情绪之间的平衡、生育行为、认知功能等。雌激素及神经递质和情感障碍的关系很复杂,主要是通过5-羟色胺系统影响心境并导致攻击行为。月经期雌激素水平升高可以降低或减少精神病发病率。临床上研究者发现精神症状在月经前或月经期加重(此期为低雌激素水平相)。在妊娠期,雌激素血清水平是常人的20~100倍,而在产后3天内便迅速下降至正常值,此时将会出现明显的心境障碍症状。绝经后女性的阿尔茨海默病、绝经后抑郁、帕金森病等精神疾患的发生日渐增多,给予雌激素替代治疗后发病有下降趋势,充分证明雌激素及其受体对脑的发育、神经递质的释放、神经元的分化、神经功能结构的维持及神经损伤的修复发挥着重要作用。
(8)皮肤:雌激素使真皮增厚,结缔组织内胶原分解减慢,使表皮增殖、弹性及血供改善。雌激素通过降低铜离子水平影响皮肤角蛋白和胶原的代谢。绝经后雌激素减少所致的胶原蛋白的丧失,水分流失还可使真皮层变薄,皮肤的分泌小体也发生萎缩。膀胱和尿道也因雌激素的减少而对外伤的易感性增加。
(9)血液系统:雌二醇是激活血小板形成的关键因素。血小板含量异常可能导致血栓的形成。
雌激素的功能主要通过与特异性受体结合而实现,目前公认的有2种受体类型,即雌激素受体α(estrogen receptor,ERα)和雌激素受体β(ERβ)。ERα由595个氨基酸组成,分子量为55kDa,其编码的基因位于6号染色体的6q5.1区;其半衰期为4~7小时,是一种快速周转蛋白。1996年从人睾丸组织克隆出的ER被称为ERβ,由530个氨基酸组成,分子量为59.21kDa,其编码的基因位于第14号染色体14q22.24;1998年报道还发现大鼠ERβ的异构体——ERβ2,其结构中插入了54个核苷酸,其中18个预计在配体结合区,编码503个氨基酸的蛋白,其与雌二醇的亲和力低于ERα或ERβ。此外,可能还有第三种雌激素受体γ(ERγ),通过ERβ基因复制得到,但由于其甾体激素特征不完全,难以确定与ERα和ERβ的相似性,因此,人类组织中的ERγ也被称为雌激素相关受体γ。G蛋白偶联雌激素膜受体(G protein-coupled estrogen receptor,GPER)是由Pietrwo和ISzego于1977年首次发现,当时认为质膜上有种能对17-雌二醇(17-E 2 )起快速反应的蛋白质,参与了早期雌激素刺激引起的表观修饰,GPER不仅与胞膜相连,也与内质网结合,目前认为雌激素与GPER的结合能力弱于其与核受体的结合。尽管已经制备了多种 Gper 敲除的小鼠,但均未发现其对雌性生殖有显著效应,其对雄性的作用似乎也不明显,其生理功能尚有待研究。但细胞膜启动的类固醇受体信号可以在几秒钟(如Ca 2+ 内流,cAMP和cGMP合成)或者几分钟(如激酶的激活)内产生信号通路蛋白的翻译后修饰效应。该效应有时会与其核受体产生的效应协同,以增强或削弱某些基因的表达,从而实现对多种器官功能活动的调节。
ERα和ERβ两种受体是受不同的基因编码,但结构上却高度同源的蛋白。其DNA结合区各有83、80个氨基酸,序列具有96%的同源性,激素结合区域各有250、243个氨基酸,有53%的同源性。ERα和ERβ广泛分布于全身,除生殖系统、乳腺外,心、脑、骨、消化道、肝、肺、肾等组织也有表达。
不同组织中,雌激素受体分布有所不同。①生殖系统:在女性,卵巢间质主要表达ERα。而颗粒细胞和黄体细胞主要表达ERβ。输卵管、子宫、阴道上皮主要表达ERα,ERβ表达微弱。男性睾丸、前列腺主要表达ERβ。敲除 ERα 基因的小鼠,子宫乳腺不发育,卵巢囊性变、不育。敲除 ERβ 基因的小鼠,子宫形态正常,生育力低下。②乳腺:正常乳腺组织ERα的表达远高于ERβ。③心血管系统:血管平滑肌主要表达ERβ,而且女性高于男性。ERα表达微弱。女性心肌ERα的表达活动高于男性,ERβ表达无性别差异。④脑:大脑皮质、小脑、海马回主要表达ERβ。在下丘脑中,弓状核和腹正中核主要表达ERα。室旁核和室上核主要表达ERβ,而视前区和终纹两者都表达。⑤骨:青少年软骨细胞、活跃的成骨细胞和干骺端主要表达ERα。成年人骨皮质的成骨细胞和骨膜表面的骨细胞中主要表达ERα,骨松质的成骨细胞和骨细胞主要表达ERβ。两种雌激素受体与不同的雌激素之间的亲和力也不同。
雌激素的作用机制如下:①经典的基因途径,是雌激素的主要作用途径,即雌激素进入细胞内,转入细胞核与受体结合,引起雌激素应答基因的转录激活;②通过基因途径的信号传导新模式;③胞膜受体相关信号通路的激活。
雌激素通过基因途径的信号传导新模式为:雌激素与受体结合,受体构型改变而被激活,形成同型或异型二聚体;首先与细胞内辅助调节因子(结合器)形成复合物,此复合物与雌激素反应元件(estrogen response element,ERE)或与其他转录因子结合,启动或抑制转录,从而产生效应。这一过程一般需要数小时或数天。其具体过程如下:
1)受体的激活:①配体依赖性激活:为经典的作用途径。配体通过与受体的配体结合区结合,使受体构型变化而被活化。ERB和EBB都有2个区域为转录激活功能1区和2区(activation function 1 and 2,AF1和AF2),可通过AF1和AF2协同或独立调控雌激素应答基因的转录不同组织ER的AF1、AF2活性不同,对转录过程的影响也可不同。不同配体可选择性地刺激或抑制两种受体的AF1或AF2,各展示出不同的活性。②非配体依赖性激活:近年研究表明,ER也可以不依赖特异配体而被其他不同的信号激活。如EGF、1GF-1、TGF-α等均可激活ER。研究发现AF缺失的ER变异体可被E 2 激活,但不被EGF或IGF-1激活;相反,缺失配体结合区的ER则可被EGF或IGF-1激活,而不被E 2 激活提示EGF、TNF-α、IGF-1等生长因子必须通过AF通路激活ER。
有报道RGF通过使ER磷酸化而激活。研究发现ER的A/B区的Ser118是ER对EGF反应的重要位点,EGF通过诱导MAPK级联反应使Ser118磷酸化,从而活化AF1,导致ER激活。另有学者发现:EGR或IGF-1与雌二醇(E 2 )共同作用比单独一种因子引起 ER 靶基因表达更强,提示其与雌激素有协同作用,而且E 2 存在时,EF或IGF-1可不通过AF激活ER,说明生长因子信号通路可作用于ER的不同功能区。
2)激素受体复合物的二聚体化:被激活的ERα和ERβ可形成同型或异型二聚体。受体的二聚体化可能涉及DNA结构的改变从而增加复物的稳定性,也可能两个ER分子相互作用以获得与ERE更高的亲和力。
3)细胞内辅助调节因子:又称为转中介因子或接合器。所有的细胞内都具有辅助调节因子,分为辅助激活因子和辅助阻遏因子两类。不同细胞中其浓度可不同。受体未被激素活化时,辅助阻遏因子多;反之,受体经激素活化后,辅助激活因子增多。它们具有加强或压制受体与促进子、SRE结合的作用。受体激活二聚化后,构型发生改变,为辅助调节因子提供结合位点。有学者认为激素受体通过这种中介因子搭桥,同结合在TATA框的转录起始复合体相互作用,共同调节转录水平。这些因子一般存在多个受体作用部位。可能有助于EB发挥不同的调控作用。已确认的能与ER的AF2相互作用的辅助激活因子有SRC-1、CBP等;已确认的辅助阻遏因子有NCOR1和SMRT。
SRC属于160kDa的辅助激活基因家族,有两种异构体SRC-1a和STRC-1e,该家族成员均有相同的高保守序列,其结合位点为ER-LBD4个螺旋区的氨基酸形成的疏水结构。研究发现配体结合ER后与SRC-1结合,可促进AF1和AF2的协同作用。在无配体时,SBC可与其他激活因子协同作用,加强或减弱ER的转录激活功能。
4)受体介导的转录激活位点:经典的雌激素反应元件由两个反向的6个核苷酸重复序列组成。有研究提示:不同的配体通过ERα与AP1反应元件结合,如17-雌二醇及他莫昔芬能充分激活靶基因的转录,而雷洛芬只能部分激活靶基因的转录;相反,不同的配体通过EBB与AP1反应元件结合,如17-雌二醇能抑制靶基因的转录,ICI-164 384却能激活靶基因。
此外,雌激素还能与膜受体结合,促使G蛋白的Gαβγ结构解离,解离后的Gα-GTPase亚基参与调节离子通道和磷脂酶C及腺苷酸环化酶活性,而Gβγ结构也参与激活下游信号。因此,通过雌二醇与膜受体的结合,引起细胞内第二信使cAMP和cGMP的合成,随后激活PI3K和MAPK信号蛋白,从而引起雌激素刺激引起的一系列表观修饰。
综上所述,雌激素和通过多条途径在机体内发挥高度复杂性的调控作用,具有显著的组织/细胞特异性。
2.雄激素 卵巢中的卵泡膜细胞是卵巢合成和分泌雄激素(主要是雄烯二酮)的主要部位,卵巢分泌的雄烯二酮约占育龄期女性体内雄烯二酮总量的50%。卵巢间质细胞和门细胞主要合成与分泌睾酮,其分泌量约占育龄期女性体内睾酮总生成量的25%。睾酮主要以葡糖醛酸盐的形式经尿排出,双氢睾酮(dihydrotestosterone,DHT)在体内转换成3α、3β-雄烷二醇及其葡糖醛酸盐,再由肾排出。
雄激素可能有两种受体:①睾酮受体:存在于中肾管系、肌肉、脑、骨髓、睾丸生精上皮等。在性分化时,睾酮能促进男胎内生殖器的形成,青春发育期,睾酮调节男性促性腺激素的分泌和睾丸的生精功能。②双氢睾酮受体:位于皮肤毛囊、皮脂腺、阴蒂、男性外生殖器及前列腺。在性分化期双氢睾酮促进男胎外生殖器及前列腺的发育;青春发育期与性毛生长、皮脂腺分泌、男性外生殖器的发育有关。
而女性体内,雄激素的功能主要有:①生殖系统:是合成雌激素的前体物质,也是维持女性生殖功能的重要激素,能促进阴毛、腋毛生长,促使阴蒂、阴阜和阴唇的发育。雄激素过多会拮抗雌激素作用,减缓子宫及子宫内膜生长和增殖,抑制阴道上皮的增生和角化,对维持女性的性欲非常重要。雄激素过多可能影响卵泡生长发育和排卵,导致月经失调。②代谢:雄激素促进蛋白质合成和肌肉生长;促进骨髓造血,刺激骨髓中红细胞增生,促进青春期少年肌细胞生长和骨骼的发育,使青春后期骨骺关闭,生长停止。③血管系统:DHT调节雌二醇水平,并且雄激素在改变脑血管张力、血管内皮功能、氧化应激和炎症反应方面有重要意义。研究发现孕产妇血清睾酮浓度的增加与异常临床表现有关,由此引起的妊娠高血压与NO介导的血管舒张功能有关。血清睾酮浓度的增加可能诱发血阻力增加,这与妊娠高血压有一定的关系。
雄激素可通过三种形式发挥作用:①在细胞内将睾酮转化为双氢睾酮;②睾酮本身发挥作用;③在细胞内将睾酮转化为雌二醇。睾酮直接作用的靶器官是中肾管的衍生结构,而毛囊、尿生殖窦和尿生殖结节的衍生结构则需要先将睾酮转化为双氢睾酮。
雄激素受体同孕激素受体类似,也存在全长的B型和较短的A型两种受体。雄激素受体DNA结合区中的氨基酸序列与孕激素受体氨基酸序列相似。对分离的人卵泡膜细胞的研究表明,卵泡膜细胞是卵泡雄激素的主要来源。卵泡膜层表达STAR、P450SCC、P450C17和2型3β-羟甾类脱氢酶,均受黄体生成素的调节。相比之下,分离培养的人颗粒细胞产生的雄激素可以忽略不计,无论是否添加促性腺激素。除了作为雌激素生产的底物外,雄激素对促进灵长类动物卵巢中初级卵泡的累积和卵泡的存活有作用。
此外,雄激素受体与细胞增殖标志物Ki-67呈正相关,与细胞凋亡呈负相关。这些观察结果与雄激素具有促进卵泡闭锁的观点形成了对比,这一概念主要来源于对啮齿类动物卵巢的研究,在某些系统中,雄激素在体外阻止颗粒细胞增殖并促进卵泡闭锁。例如,在缺乏促性腺激素的情况下,雄激素可引起垂体切除未成熟大鼠的滤泡闭锁和对抗雌激素相关的卵巢重量增加。同样地,用5α-二氢睾酮治疗可使FSH在颗粒细胞中诱导黄体生成受体并抑制颗粒细胞增殖。对绒猴的研究表明,雄激素对体外颗粒细胞功能的调节呈现显著的阶段性。雄激素增强了FSH刺激芳香化酶的表达和孕酮的产生,同时抑制了hCG刺激的芳香化酶活性和大的排卵前卵泡细胞中孕激素的合成。虽然研究发现卵泡液含较高水平的5α-二氢睾酮和较低水平的雌二醇是闭锁卵泡的特征。然而,激素水平的变化是事件的结果而不是闭锁的原因,有报道称,卵泡中降解的高浓度的雄激素(如5α-二氢睾酮)可作为颗粒细胞芳香化酶活性的竞争性抑制剂。因此,多囊卵巢综合征患者的卵泡比正常卵巢的卵泡具有更大的5α-还原酶活性。综上所述,雄激素可能通过雄激素受体以及非受体介导的机制,以阶段依赖的方式对卵泡生长和功能产生正负两方面的调节。
3.孕激素 卵巢分泌的孕激素包括孕酮、17α-羟孕酮,主要由颗粒黄体细胞及卵泡膜黄体细胞生成。由于卵泡内无血管供应,颗粒细胞缺乏合成孕酮所必需的低密度脂蛋白胆固醇,只有在黄素化后,有直接的血液供应,才能得到低密度脂蛋白胆固醇,合成与分泌孕激素。育龄期女性体内孕酮的含量在卵泡期为2mg/d。一般排卵后l周,即月经周期的第20天左右黄体发育成熟,孕激素分泌量达最高峰,达到25mg/d,随黄体萎缩分泌量逐渐下降,至月经来潮时,恢复到排卵前的水平。临床常用测定尿中孕二醇作为诊断有无排卵的一个重要指标。孕激素的代谢产物为孕二醇;经肾排出体外。
孕激素通常在雌激素作用的基础上发挥作用。孕激素能抑制雌激素受体的补充,促进雌二醇代谢,因此有抗雌激素的作用。孕激素也能抑制其自身受体的生成:①生殖系统:孕激素抑制子宫肌层收缩,降低子宫平滑肌兴奋性及其对缩宫素的敏感性,有利于胚胎及胎儿在宫内生长发育。对抗雌激素的内膜增殖作用,使增生期子宫内膜转化成分泌期子宫内膜。调节毛细血管舒缩功能,提高子宫内膜血流,增加基质水肿和间质蜕膜样变,有利于孕卵的着床及发育,抑制母体对胎儿的免疫反应,有利于妊娠的维持,能促进和维持黄体功能。使宫颈口闭合,抑制宫颈腺体分泌,黏液分泌减少,性状变黏稠,不利于精子穿透。抑制输卵管平滑肌节律性收缩频率和振幅,抑制输卵管上皮纤毛生长,调节孕卵的运行。使阴道上皮角化减少,中层细胞增多,加快阴道上皮细胞脱落。②下丘脑和垂体:孕激素在月经中期具有增强雌激素对垂体LH排卵峰释放的正反馈作用;在黄体期对下丘脑、垂体有负反馈作用,抑制促性腺激素分泌。③乳腺:在雌激素作用的基础上,孕激素促使腺泡发育,妊娠期高浓度的孕激素进一步促进乳腺发育,为泌乳作好准备;大量孕激素抑制乳汁分泌。生育年龄女性乳腺增生随月经周期发生周期性变化,乳腺上皮增生与孕激素水平显著相关,黄体期增生明显。④代谢:影响蛋白、脂肪及碳水化合物的代谢,促进蛋白质的分解。在肾小管竞争结合醛固酮受体,促进水钠排泄。⑤神经系统:对下丘脑体温调节中枢有兴奋作用,可使基础体温在排卵后上升0.3~0.5℃,临床上可作为判断是否排卵及排卵日期的标志之一。此外,研究表明孕激素对脑损伤的神经再生和修复以及髓鞘修复均有积极意义。⑥呼吸系统:孕酮刺激呼吸。在黄体期,女性肺泡PCO 2 比男性低即是孕酮的作用。孕酮有改善肺通气、升高PaO 2 及降低PaCO 2 的作用。⑦皮肤:孕酮可导致一种罕见的皮炎:自身免疫性孕酮皮炎(autoimmune progesterone dermatitis,APD)。在月经周期的黄体期孕激素水平升高时,出现皮疹临床症状有多形性红斑、湿疹、荨麻疹、血管神经性水肿,甚至出现酮诱发的过敏性休克等。
孕激素受体存在于细胞核内,是单一多肽链,其结构、转录活化的调节基质与雌激素受体相似。不同的是A/B区有2个转录激活功能区。孕激素受体主要分为两个蛋白亚型:PR-A和PR-B。两种受体来源于同一编码基因,由于不同的启动子和不同转录起始点所致,PR-B是全长形式,由934个氨基酸残基组成,分子量116kDa;PR-A由771个氨基酸残基组成,分子量94kDa。PR-B和PR-A相比有一个氨基端延长区,此区内有一个AF区,这决定了PR-B活化的特异性。PR-B和PR-A与孕酮结合后形成二聚体,参与基因表达的调控。当细胞内A和B两型受体呈等摩尔数表达时,A和B型受体可形成三种二聚体:A:A、A:B、B:B。特殊条件下靶细胞内A和B两型受体表达的差别会导致二聚体组成的变化,从而使细胞对孕酮的反应有所不同。此外尚存在一种PR-C亚型,是PR-B的N末端截短的形式,分子量60kDa。PR-C与孕酮的解离系数比PR-A和PR-B高约5倍,可与PR-B形成异二聚体,干扰同二聚体的形成。PR在卵巢、子宫、乳腺、神经系统和胸腺中表达。大量证据表明PR-A、PR-B有不同的功能。在体外培养的细胞中,当激活剂与PR-B结合后,表现为强的转录激活作用,而在多种细胞类型中PR-A是非活化的;当拮抗剂与PR-A结合后是失活形式时,而拮抗剂结合的PR-B可通过胞内磷酸化途径的改变而成为活化的形式;结合拮抗剂的两型受体都有抑制雌激素受体的作用。
此外,孕激素受体还能通过配体非依赖的途径被激活,生长因子类和多巴胺等可提高胞内激酶活性,可激活胞内的磷酸化途径,从而使PR磷酸化而激活。
颗粒细胞和膜间质细胞一样,在黄体生成激增后,为孕激素的合成作了充分的准备。这种激增触发了编码 STAR 、 P450scc 和2型3β-羟甾类脱氢酶(有效合成孕酮所需的三联体蛋白)基因的表达。如前所述,排卵需要排卵前卵泡产生孕酮,它也可能在调节黄体功能中起作用。3β-羟甾类脱氢酶抑制剂对卵巢孕酮产生的药理学阻断表明,孕酮对黄体生成细胞具有抗凋亡和促分化作用,并维持黄体功能。孕激素受体拮抗剂米非司酮和HRP2000抑制人颗粒黄体细胞hCG刺激的孕激素和松弛素的分泌。孕激素受体(A和B型)存在于恒河猴和人的黄体中,孕激素受体的mRNA从早期到中期逐渐增加。黄体早期至晚期,孕酮受体B与孕酮受体A的比值增加。孕酮受体拮抗剂对黄体细胞甾体生成的作用可能是这些核受体调节的转录改变的反映。
抑制素是TGF-β蛋白超家族的成员,是分子量为32kDa的异二聚体糖蛋白,分别由α亚基(18kDa)和β亚基(12kDa)通过二硫键连接组成。其α亚基相同,但有不同的β亚基,分别称为β A 和β B 。αβ A 和αβ B 异二聚体分别命名为抑制素A和B。抑制素的主要产生部位是性腺。在卵巢中的主要来源是颗粒细胞。抑制素的内分泌功能是抑制垂体促性腺激素的产生,但同时也参与多种生物学功能的调节,从胚胎发育的早期阶段到最终分化的细胞和组织中均发挥高度特异性的调节功能。在体外,它对促黄体激素和胰岛素样生长因子刺激的卵泡膜细胞产生的雄激素起协同作用。尽管抑制素的两种亚型具有相似的生物学特性,但它们在卵泡中的形成时期不同。抑制素B主要在卵泡早期分泌,在卵泡中期分泌下降,在LH峰以后检测不到。在卵泡发育的前半周期,抑制素A的浓度较低,但在卵泡发育中期和黄体期,其浓度升高。抑制素A的分泌受促性腺激素的调节,但抑制素B的产生明显不受调节。在不同大小的卵泡上进行的测量表明,抑制素A和B表达存在显著的差异性,在< 6mm大小的卵泡中,抑制素A的含量随着卵泡大小的增加而增加。相反,抑制素B的含量与卵泡大小或成熟度无关。尽管抑制素是卵巢的分泌产物,但它也在卵巢内发挥作用。如前所述,它增加了卵泡膜细胞产生的促黄体激素和胰岛素样生长因子刺激的雄激素。
由抑制素的两个β亚单位(β A β B 、β A β A 或β B β B )组成的同源二聚体,因其刺激培养的垂体细胞分泌FSH而得名。激活素A的水平在月经中期和晚期黄体期/早期卵泡期较高,而在妊娠期则明显升高。在卵泡液中激活素A的浓度与卵泡大小或成熟度无关。然而,由于抑制素A水平随着卵泡大小和成熟度的增加而增加,卵泡发育的特点是从以激活素为主的环境转变为以抑制素A为主的环境。激活素对卵泡成熟和颗粒细胞功能具有阶段依赖性作用。激活素促进未成熟颗粒细胞增殖,并诱导FSH受体和芳香化酶的表达。更成熟的颗粒细胞在激活素作用下分化。颗粒细胞来源的激活素增强了由FSH支持的颗粒细胞中LH受体的表达和功能。在卵泡膜细胞中,激活素与抑制素的作用相反,抑制促黄体生成素刺激的雄激素合成。在人类颗粒细胞中,激活素抑制基础水平的以及由促性腺激素刺激产生的孕酮和雌激素。卵母细胞表达激活素受体,这可能是颗粒细胞通过旁分泌作用调节卵母细胞发育的途径。有证据表明,激活素能促进卵母细胞成熟。小鼠基因“敲入”研究表明,激活素A和B在卵巢中不具有重叠功能,β A 亚单位对正常卵泡发育是必要的。有学者从猪卵泡液中分离得到了卵泡抑素,并根据其抑制FSH活性的基础上命名。由于C末端的可变剪接、糖基化和蛋白水解加工,卵泡抑素以多种形式(315和288个氨基酸)存在。卵泡抑素由小窦和排卵前卵泡产生,且近乎不可逆地结合激活素并有效地中和其活性。随着卵泡大小或成熟度的不同,卵泡液中的游离卵泡抑素水平也会发生变化。卵泡抑素在整个月经周期内的循环浓度相对恒定。在转基因小鼠中,卵泡抑素的过度表达导致第二阶段的卵泡成熟停滞,证实了激活素在卵泡成熟中的重要作用。
AMH是TGF-β超家族的成员。除了在雄性分化过程中诱导米勒管变性的既定功能外,还在成年卵巢中发挥重要调节作用。AMH主要由小卵泡的颗粒细胞产生,通过两个丝氨酸/苏氨酸激酶受体传递信号,其中Ⅱ型为特异性受体,Ⅰ型受体与BMP家族共享。缺乏AMH的雌性小鼠表现为卵泡的加速衰竭,反映了AMH对始基卵泡向生长池中募集的抑制作用,以及导致生长卵泡对FSH的反应性减弱。因此,AMH可抑制FSH诱导的体外窦前卵泡生长。目前AMH被认为是卵巢储备的可靠标志物,可以帮助预测早期卵巢卵泡的丢失和更年期发生。AMH水平也反映了侵入性妇科手术或促性腺激素治疗,如化疗对卵巢储备的影响。此外,AMH参与某些疾病的诊断,如颗粒细胞瘤或多囊卵巢综合征(polycystic ovarian syndrome,PCOS)。然而,不建议AMH作为预测辅助生殖的胚胎植入存活率、妊娠率和活产率等的独立指标,且检测水平及可靠性仍存在争议。此外,近期还发现AMH除了可预测卵巢功能外,调节下丘脑、垂体发育及抗肿瘤的作用。
通过分析人体颗粒黄体细胞和表面上皮细胞中的RNA,已证实GnRH-Ⅰ、GnRH-Ⅱ和GnRH受体的表达,尽管其表达水平分别低于下丘脑和垂体中的水平。几项体外研究已经阐明了GnRH-Ⅰ和GnRH-Ⅱ对抑制甾体生成和卵巢表面上皮细胞增殖剂量依赖性作用。以上研究表明人卵巢内GnRH系统的存在。在啮齿类动物卵巢中该系统具有更大的作用。GnRH受体的存在也增加了卵巢作为GnRH激动剂和拮抗剂的靶点的可能性。然而,大多数临床研究表明其对卵巢功能的影响是次要的,但也为GnRH-a对抗医源性卵巢衰竭提供理论支持。
松弛素是一种可能促进子宫内膜蜕膜化和抑制子宫肌层收缩作用的激素,由黄体的大黄体细胞产生,从黄体早期到晚期逐渐积累。松弛素在妊娠的前三个月达到最高循环水平,然后下降约20%,并在整个妊娠期间保持恒定。胰岛素样肽3(INSL3)过去被称为松弛素样因子,是松弛素家族中的一员。它最初被认为是睾丸间质细胞分泌的主要产物,是睾丸间质细胞功能的重要生物标志物。然而,INSL3也由生长中的窦状卵泡的卵泡膜细胞产生。在卵泡内,INSL3通过其G蛋白偶联受体RXFP2,以自分泌/旁分泌的方式,调节和驱动类固醇前体雄烯二酮的产生并由颗粒细胞转化为雌激素。由此形成了一个正反馈循环,促进卵泡膜细胞表达更多的INSL3。这与卵泡生成和LH峰相抵消。因此,卵泡膜细胞INSL3-RXFP2系统的活性,有效地决定了卵泡期窦状卵泡中雌二醇的产生。对基因敲除小鼠的研究证实,雌性小鼠体内INSL3或其受体的缺失导致部分不孕,卵泡数量、排卵和产仔数减少。INSL3将根据卵泡的数量和健康状况分阶段地分泌到血液循环中,在循环中可作为一个监测窦状卵泡生长状况的有利生物标志物,因此,在多囊卵巢综合征中分泌增加,在POF女性中减少。作为一种内分泌因子,INSL3还可能影响骨代谢和肾功能。此外有研究证实,利用INSL3或其类似物作为激素替代治疗、监测或影响体外受精方案的辅助分子也被证明是有价值的,具有较好的前景。
生殖系统以经典的内分泌模式进行运作,下丘脑产生以脉冲形式释放的促性腺激素释放激素(gonadotropin-releasing hormone,GnRH),是月经周期的始动者。GnRH调节垂体前叶内合成并释放促性腺激素到血液循环中,包括FSH和黄体生成素(luteinizing hormone,LH)。FSH和LH刺激卵泡的发育和排卵及黄体形成,并调节卵巢激素的分泌。此外,卵巢对下丘脑、垂体的反馈调节及局部的旁分泌调节和其他内分泌系统,包括肾上腺、甲状腺等,也对卵巢功能起重要调节作用。
卵巢类固醇通过其对作用于下丘脑中GnRH神经元的上游的KNDy神经元的作用影响GnRH分泌的幅度和/或频率。kisspeptin/神经激肽B/强啡肽神经元位于动物的下丘脑弓状核和人类的漏斗状核,因其共同表达kisspeptin、神经激肽B和强啡肽而被命名为KNDy神经元。此外,卵巢类固醇和抑制素直接作用于垂体,FSH分泌的负反馈抑制对于人类生殖周期中单个成熟卵母细胞的发育至关重要。除了负反馈抑制之外,排卵过程中所必需的排卵前LH峰则依赖于雌激素的正反馈,使得月经周期周而复始的规律发生。
1979年,促黄体素释放激素(luteinizing hormone releasing hormone,LHRH)被科学家分离、描述和合成。由于这一十肽化合物在物种繁殖中占据的核心地位,使得分离出这一肽类的科学家Drs.Schally和Guillemin获得了1977年的诺贝尔生理学或医学奖。后来预计将发现分别单独促进LH和FSH释放的激素,然而,随后的研究提供了LH和FSH都对LHRH做出反应性释放的证据。因此,最初称为LHRH的十肽化合物现在已被统一改称为GnRH。
1)GnRH的编码基因:
哺乳动物,包括人类,同时表达一种以上分子形式的GnRH(表2-4)。编码GnRH的基因位于8号染色体短臂上,有4个外显子,GnRH通过其受体发挥作用,编码受体的基因作用于4号染色体上。编码GnRH-Ⅱ的基因位于20号染色体短臂上,通过其独特的受体发挥作用,该受体在1号染色体上编码,受体可能在低等动物物种的繁殖行为中发挥作用。它是动物体外和体内模型的有效刺激物,但在人体中作用尚不清楚。在一些低等动物物种有证据表明,在七鳃鳗中分离的GnRH-Ⅲ可能具有优先的FSH释放特性。然而,人类基因组中尚未发现GnRH-Ⅲ的共有序列,目前认为GnRH-Ⅲ在人类生殖中可能不发挥作用。
表2-4 哺乳动物GnRH-Ⅰ、GnRH-Ⅱ、GnRH-Ⅲ的氨基酸序列

2)GnRH/GnRHR系统基本特征:
GnRH前体基因GNRH-Ⅰ位于8号染色体上,在哺乳动物中,线性十肽的终产物是由视前下丘脑的89个氨基酸前体激素合成的。它是下丘脑性腺轴各种调节机制的靶点。
GnRH神经元在嗅觉基板中分化,穿过筛状板进入前脑,并迁移到内侧基底下丘脑,在那里它们与垂体门脉系统建立连接,作为下丘脑结节系统的一部分。在人类,大脑区域内约有7 000个表达GnRH的神经元与促性腺激素调节相关。与分泌其他下丘脑释放因子的神经元不同,GnRH神经元不存在于特定的核中,而是散布在整个内侧基底下丘脑中,另外还有分散在视前区的神经元。
GnRH和GnRHR已经被证实在不同的物种(包括人类)的卵巢水平表达。如前所述,对于垂体GnRHR,根据卵泡发育阶段这些结合位点的表达也遵循卵巢水平的动态模式。目前已经证实,大鼠GnRHR水平在早期窦卵泡中表达水平较低,且在卵泡生长的过程中逐渐增加,在成熟卵泡(又称赫拉夫卵泡,Graafian follicle)和闭锁卵泡的颗粒细胞中高度表达。在人类的研究中,GnRH和GnRHR在发育早期阶段的卵泡中都不表达,而是存在于成熟卵泡的颗粒细胞层以及颗粒-黄体细胞中。卵巢GnRHR据报道是对应于垂体水平的受体,并且已经在人类卵巢细胞中证实了GnRH-Ⅱ的存在,支持了这种形式也可能参与调控卵巢功能的观点。综上所述,这些实验结果支持GnRH/GnRHR系统更可能参与卵泡发育/闭锁和黄体化/黄体溶解的过程。颗粒细胞和卵泡膜细胞内层细胞中发生的细胞凋亡在卵泡发育中起到关键作用。在初级卵泡中尚未观察到凋亡的颗粒细胞,而窦卵泡中颗粒细胞凋亡细胞的数量增加。在窦卵泡闭锁的早期阶段,颗粒细胞中存在细胞凋亡,而在这些闭锁晚期的卵泡膜细胞中的细胞凋亡更加显而易见。基于GnRH对卵泡细胞活力的影响,有人提出,GnRH/GnRHR系统可能参与了卵泡闭锁的促凋亡过程。在大鼠和猪颗粒细胞中,GnRH减少了DNA的合成并诱导来自较小的卵泡中的细胞凋亡。人颗粒细胞中进行的研究报道了类似的观察结果。获得的结果表明,GnRH下调人颗粒细胞的增殖,通过干扰IGF-1/AKT信号通路并诱导Caspase 8,3,7直接触发外源性凋亡途径。总之,通过一系列观察结果强烈支持GnRH/GnRHR系统在调节卵泡发育和闭锁中的重要作用。
3)GnRH激动剂(GnRH agonist, GnRH-a)的作用:
卵巢的正常发育和功能与激素的正确合成和卵泡成熟相关,在人类颗粒细胞中,GnRH-a可以提高芳香化酶的表达,从而刺激雌激素的产生;GnRH-a可增加AMH的表达,参与卵泡的发生过程。GnRH同样诱导与卵母细胞成熟和排卵时卵泡破裂相关的基因的表达,包括孕激素受体基因和血纤维蛋白溶酶原活化因子。此外,在颗粒细胞中,GnRH-a可提高色素上皮衍生因子(pigment epitheliumderived factor,PEDF,一种有效的抗血管生成因子)与VEGF的比值,与人绒毛膜促性腺激素(human chorionic gonadotropin,hCG)相反。基于该项观察结果,有研究者提出,与辅助生殖方案中的hCG相比,这种GnRH相关的抗血管生成作用可能与GnRH-a获得的卵巢过度刺激综合征(ovarian hyperstimulation syndrome,OHSS)风险降低有关。另外,由GnRH-a诱导的抗血管生成环境可能损害黄体的功能,而正常的黄体功能依赖于正确的血管生成过程。
已有研究报道GnRH/GnRHR系统也可能参与黄体化和黄体溶解的过程,特别是在人颗粒-黄体细胞中,GnRH-a降低雌激素受体ERα和ERβ的表达。GnRH-a在黄体中诱导黄体溶解,这与基质金属蛋白酶(如MMP2)和1型膜MMP的刺激有关,导致发育中的黄体再生和细胞外基质的重塑。对于GnRH-Ⅱ也报道了类似的情况。具体说,GnRH-Ⅱ可以抵消来自人颗粒-黄体细胞中hCG诱导的孕酮分泌。在这些细胞中,GnRH-Ⅱ诱导FSH和LH受体的下调。因此,卵巢中局部表达的GnRH(和GnRH-Ⅱ)/GnRHR系统参与卵泡发育/闭锁和黄体功能的调节。关于GnRH释放的调控以及其类似物在治疗方面用途的最新数据为该领域提供了一系列新观点。GnRH/GnRHR系统在雌性生殖轴中表达,来自下丘脑神经元的GnRH的脉冲释放由一系列正在研究的神经肽精细调节,将成为未来治疗生殖系统疾病创新治疗方法的新的靶标。GnRHR在卵巢水平的功能尚未完全阐明,但其可能为卵巢功能障碍提供新的干预手段。最后,应用GnRH-a来保护年轻女性化疗患者的卵巢功能是一个值得进一步研究的热点方向。
4)影响GnRH合成的相关基因:
对孤立性促性腺激素释放激素缺乏症(isolated gonadotropin deficiency,IGD)患者进行遗传学研究,伴随嗅觉系统破坏导致嗅觉丧失(Kallmann’s syndrome,KS)或无嗅觉丧失,也即嗅觉正常的特发性低促性腺激素性性腺功能减退症(normosmic idiopathic hypogonadotropic hypogonadism,nIHH)的发现使得我们对生殖系统复杂的神经内分泌调控的理解有了前所未有的增长。现已经在这一罕见病群体中发现了超过35个基因的突变。
通过在细胞和动物实验中验证它们的功能可以将其大致分为四组(表2-5)。
表2-5 导致IGD的基因分类与列表

续表

5)GnRH的脉冲样释放:
将GnRH脉冲样分泌释放到垂体门脉系统中以促进正常的促性腺激素分泌是生殖系统的一个显著特征。其脉冲频率是60~120分钟,其频率与月经周期时相有关。现已有研究表明GnRH的分泌具有脉冲样节律,该节律由弓状核内部固有的节律所决定,其频率和幅度继续严格限定在一定范围内才能发挥其调节垂体促性腺激素的合成和分泌作用,从而维持正常的月经周期。有研究证实,只有在生理节律条件下的GnRH的刺激(60~90分钟),才能引起垂体FSH和LH的生理性分泌,并有效促进卵泡的发育及雌二醇的分泌。GnRH释放频率减慢将导致无排卵和闭经;释放频率过快或持续释放,将会出现垂体促性腺激素分泌及卵泡发育的抑制,这是由于GnRH的持续刺激引起了垂体促性腺激素细胞上GnRH受体的降调节而导致垂体对GnRH失去了敏感性。分离出的GnRH神经元表现出内在的脉冲规律,但外部的影响也可以改变并调控GnRH的分泌,从而影响GnRH分泌的幅度和频率。
生殖内分泌系统是一个复杂而精细的系统,在这个系统中,GnRH不仅通过下丘脑-垂体-卵巢轴(hypothalamic-pituitary-ovarian axis,HPO axis)调节性激素的分泌,同时也接受性激素的反馈调节。然而,性激素并不能直接作用于GnRH神经元,需要通过中间介质。人们一直在寻找GnRH的启动因子及性激素反馈调节GnRH的中间介质。2007年,Goodman等首先在羊的下丘脑弓状核中发现一群神经元共表达kisspeptin、NKB和Dyn,这3种神经肽均参与生殖内分泌的调节。kisspeptin/神经激肽B/强啡肽[kisspeptin/neurokininB(NKB)/dynorphin(Dyn),KNDy神经元不仅能够激活GnRH神经元,同时还介导了性激素对GnRH的负反馈作用,从而确立了KNDy神经元在生殖内分泌系统中的重要作用。
1)kisspeptin:
肿瘤转移抑制因子kisspeptin又称亲吻素或吻素,其前体又称转移抑制素(metastin),是 kiss 基因所编码的多肽产物,属神经内分泌多肽激素。在脊椎动物中具有多个功能性配体。kisspeptin受体GPR54突变将导致小鼠丧失生殖功能,并引发了低促性腺激素性功能减退症,与此同时,敲除小鼠的 kiss 或 gpr54 均将对HPO轴功能造成严重影响,从而确立了kisspeptin/GPR54系统在脊椎动物生殖中的重要地位。而后kisspeptin/GPR54系统成为了脊椎动物生殖内分泌研究的热点。
青春期发动的特点是促性腺激素分泌的增加,性成熟并获得繁殖能力。青春期启动主要是经由GnRH神经元的激活,该过程涉及kisspeptin/GPR54信号的传导,并最终导致GnRH神经元的激活。现在已经确定kisspeptin途径是GnRH分泌的关键上游调控因子。de Roux N等在IGD患者的病例研究中发现了kisspeptin在生殖系统中的作用,该研究鉴定了编码kisspeptin受体(KISSI1R,也被称为G蛋白偶联受体54,GPR54)的基因突变。kisspeptin与下丘脑GnRH神经元上的KISS1R相结合可以激活GnRH神经元,导致GnRH大量分泌。一系列动物体内实验证明,在大鼠、小鼠和绵羊的体内添加了kisspeptin后,可以出现LH和FSH的释放,且这些促性腺激素的释放已经被证明是剂量依赖性的,加大kisspeptin肽的剂量后会导致更多的LH和FSH的释放。
表达kisspeptin的神经元位于:前房室周核(anteroventral periventricular nucleus,AVPV)、脑室周围核(periventricular nucleus,PeN)、前后视前核(anterodorsal preoptic nucleus,ADP)和弓状核(the arcuate nucleus,Arc)。kisspeptin的神经元发送投射神经元至内侧视前区(medial preoptic area,MPOA),是GnRH细胞体富集的区域。该解剖学证据也表明,kisspeptin纤维与GnRH神经元有密切的解剖学关系。
kisspeptin系统被认为是激活GnRH神经元、启动促性腺激素分泌和青春期发育的神经内分泌闸门。研究发现, KISS1R 基因失活突变可导致人类或小鼠青春期发育障碍、性腺功能减退及不孕不育。Teles等在2008年报道了世界上第1例因 KISS1R 基因功能获得性突变导致的中枢性性早熟。对该患儿进行基因检测发现, KISS1R 基因存在p.Arg386Pro突变。该突变导致kisspeptin和KISS1R结合后,细胞内信号传导通路的延长激活,进而导致对细胞内信号脱敏率的明显下降,造成信号传导通路的大量激活,促进GnRH神经元分泌大量的GnRH导致中枢性性早熟的发生。
另外,对人类和大鼠的研究已经表明,kisspeptin的结合刺激PIP2水解,Ca 2+ 动员,花生四烯酸的释放,细胞外信号调节蛋白激酶1(extracellular signal-regulated kinase 1,ERK1)、ERK2和p38 MAP激酶的磷酸化。短杆菌肽穿孔贴片的实验结果表明:约30%的GnRH神经元对青春期前雄性小鼠神经元添加的kisspeptin有反应,而成年小鼠中则有60%的GnRH神经元有反应。因为只有成年的小鼠对低剂量的kisspeptin有反应,所以从侧面证明了GnRH神经元在青春期过程中被kisspeptin发育激活。
kisspeptin/GPR54系统的发现极大地促进了人类对生殖内分泌调控的认识,其中包括青春期启动的分子定时、性类固醇激素对促性腺激素分泌的正/负反馈调控、性别二态性的发生机制、季节性繁殖的生殖内分泌调控以及能量平衡和生殖的整合。
2)Neurokinin B:
Neurokinin B(NKB)又名神经介素K(neuromedin K),与P物质、神经激肽A同属速激肽(tackyinin)家族。该家族成员拥有共同的羧基端序列:苯丙氨酸-X-甘氨酸-亮氨酸-蛋氨酸-NH2。NKB在人类中由tachychinin3基因编码( TAC3 ),该基因位于12q13-q12。目前发现3种速激肽受体,即NK1R、NK2R和NK3R,均属G蛋白偶联受体的类视紫红质家族。其中NKB和NK3R亲和力最强,NK3R由 TACR3 基因编码。NKB主要通过其受体NK3R发挥作用,是除kisspeptin外在GnRH脉冲分泌调控中起关键作用的又一因子。
通过对IGD患者的遗传学调查研究发现,NKB和NK3R参与GnRH分泌的正常调控,且刺激LH分泌,作用于GnRH神经元的上游。NKB广泛表达于中枢神经系统,人类表达NKB mRNA的神经元主要位于弓状核和下丘脑的前部。对基因敲除的小鼠进行研究证实雌激素对GnRH的负反馈调节由雌激素受体α(ERα)介导,然而GnRH神经元并不表达ERα,从侧面证明了其他神经元参与发挥调节作用。NKB在绝经后妇女的下丘脑的弓状核中基因表达升高。雌激素替代治疗不仅会降低表达NKB mRNA神经元的数量,也降低了单个细胞内的表达水平。
在卵巢切除的大鼠中 NKB 基因的表达量升高并且在雌激素替代后降低,雌激素在啮齿类动物大脑弓状核中的作用主要通过ERα介导。此外,卵巢切除可以上调小鼠 NKB 基因的表达,雌激素替代可以抑制小鼠NKB基因的高表达。在切除 ER α基因敲除的小鼠的卵巢并给予雌激素替代治疗后并不能观察到NKB被抑制的现象。因此,弓状核内的 NKB 基因表达受到雌激素负反馈调节,并且由ERα介导。
3)内源性阿片类物质/强啡肽:
强啡肽(dynorphin,Dyn)是一种内源性阿片肽,主要通过kappa阿片受体(kappa-opioid receptor,KOP)发挥生理作用。有大量证据表明,使用阿片受体拮抗剂纳洛酮在女性的研究中,内啡肽参与的传导黄体酮对脉冲样GnRH分泌呈负反馈作用。但是纳洛酮不仅与mu受体结合,而且与kappa和gamma受体结合,因此,早期的实验并不能提供特异性的证据。与kappa阿片受体结合的强啡肽现在被认为是黄体酮负反馈循环的关键因子。
kisspeptin/神经激肽B/强啡肽[kisspeptin/neurokininB(NKB)/dynorphin(Dyn),KNDy]神经元不仅能够激活GnRH神经元,同时还介导了性激素对GnRH的负反馈作用,并被认为参与了GnRH分泌的起始和终止,促使其产生了脉冲样分泌,因而确立了KNDy神经元在生殖内分泌系统中的重要作用。
4)RFamide肽:
促性腺激素抑制激素(gonadotropin inhibitory hormone,GnIH)最早在鸟类中被发现,现已经在多种动物中证实其对下丘脑生殖功能有抑制作用。在人类,RFRP-1和RFRP-3神经元向GnRH神经元发送轴突投射,RFRP被分泌到垂体门静脉系统中,其受体G蛋白偶联受体147(GPR147)则存在于GnRH神经元和下丘脑中。RFRP在下丘脑和垂体中发挥调节LH和FSH分泌的作用。目前在人类中开展的实验有限:3小时输注合成的GnIH导致绝经后妇女对LH分泌的适度抑制。需要进一步的研究来确定RFRP在女性中的生理学功能和潜在治疗作用。
LH和FSH在促性腺激素细胞中合成,其占垂体中细胞的7%~15%,大鼠的免疫组化表明大约70%的促性腺激素细胞会染上LH和FSH,而其余染上LH或FSH的数量大致相等。当动物接近动情前期促性腺激素激增的那天,单激素细胞开始表达β-LH和β-FSH,另外表达促生长激素(growth hormone,GH)的细胞群也表达促性腺激素亚基。完整的促性腺激素的生物合成涉及:①β-LH、β-FSH和常见促性腺激素α-亚基的翻译;②翻译后修改和折叠;③β和α亚基的组合;④当LH和FSH穿过高尔基复合体时,LH和FSH上的寡糖残基的修饰。FSH以分泌为主,几乎没有储存。相反,LH被包装成颗粒并储存。而后由于促性腺激素细胞的GnRH刺激,分泌LH和FSH。因碳水化合物结构和电荷不同,LH和FSH具有多种亚型,在垂体和血清中共存。LH和FSH更碱的形式会使得产生体外活性更强,但是循环中的半衰期更短,而对于少碱的亚型则相反。若FSH上存在更多的唾液酸残基会延长其半衰期,而LH上磺化的N-乙酰基-半乳糖胺(GalNAc)天冬酰胺连接的寡糖的数量越多,则因与特异性肝受体的结合而更快地清除相关。LH和FSH的磺化和唾液酸化在月经周期和无性腺类固醇的情况下有所不同;绝经后妇女LH和FSH的唾液酸化形式占优势,LH和FSH中磺化和唾液酸化残基的数量与女性激素清除率密切相关。
尽管LH和FSH都是从促性腺激素中常见的细胞类型分泌的,但两者在控制卵巢生理学功能方面有着显著的不同,会在正常的月经周期中以不同的分泌模式反映出来。通过GnRH刺激模式的差异来控制LH和FSH的合成和分泌,激活素/卵泡抑制素系统对FSH合成的优先控制以及卵巢类固醇的差异性反馈,实现对LH和FSH的不同调控。理解LH和FSH分泌的控制对于我们理解月经周期的动态变化至关重要。
1)促性腺激素释放激素对LH和FSH的差异性调控:
在GnRH的急速刺激下,FSH与LH一起分泌,但GnRH在FSH合成的总体调控中的相对作用远小于LH。利用GnRH受体的特异性阻断剂阻断GnRH受体信号,对LH分泌达到90%的抑制作用,但对FSH仅有40%~60%的抑制作用。LH和FSH的合成和分泌受GnRH刺激的幅度和频率的不同控制。LH对GnRH剂量的增加具有较高的反应性,而FSH对GnRH剂量相对不敏感。GnRH的生理性脉冲频率调控促性腺激素的合成与分泌,然而不同的频率对于LH和FSH有不同的影响。
GnRH刺激的频率缓慢有利于体外FSH的合成和分泌,并且与人类研究者性腺低反馈导致FSH的增加相关。在GnRH缺乏的男性和女性中,GnRH刺激频率的增加提高了LH的平均水平,但FSH水平影响无明显的变化。而对于PCOS患者,垂体脉冲的GnRH刺激频率增加,会导致LH与FSH的比率增加,为PCOS的一个病理生理学特点。GnRH脉冲频率对GnRH受体数目的影响也是LH和FSH分泌频率调节的基础。此外,增加的GnRH脉冲频率增加卵泡抑素,会降低激活素对FSH合成的刺激。尽管GnRH脉冲频率的增加会提高LH的合成与均值水平,但LH脉冲幅度与GnRH脉冲频率呈负相关。对GnRH缺陷的男性与女性的研究表明,较慢的GnRH脉冲频率与LH较高的脉冲幅度相关,而较快的频率与LH脉冲幅度对GnRH的反应性下降相关。脉冲频率比生理环境中更快的LH振幅的降低可能是垂体脱敏的最早迹象,其与GnRH的连续输注或者使用GnRH激动剂有关。
2)促性腺激素的自分泌/旁分泌调节:
近年来,在哺乳动物中存在一类亲水性的非类固醇物质,即抑制素(inhibin,INH)、激活素(activin,Act)和卵泡抑素(follistatin,FS)。由垂体细胞和卵巢颗粒细胞生成,通过内分泌机制调节FSH的分泌。
激活素和抑制素是TGF-β超家族的成员,由α和β两个亚基组成。有研究表明,生物体内存在的β亚基有5种活性形式:βA、βB、βC、βD和βE,并且这5种亚基约有50%的氨基酸同源性。
目前研究较多的抑制素是由α亚基与βA和βB亚基连接形成抑制素A(α-βA)和抑制素B(α-βB)两种,两者均可以抑制垂体FSH的合成和分泌,抑制素A主要由优势卵泡和黄体分泌,抑制素B主要由中小窦状卵泡分泌。抑制素通过与β聚糖结合并隔绝Ⅱ型激活素受体,作为刺激FSH的激活素的特异性拮抗剂。尽管抑制素是在垂体中产生的,但来自卵巢的循环抑制素在FSH的负反馈调控中起着更大的作用。此外,骨形态发生蛋白BMP-6和BMP-7能够调节促性腺激素细胞中FSH的合成,表明其他系统也可能参与FSH的调控。
激活素由抑制素β亚基的βA和βB通过聚合作用形成活化素A(β A -β A )、AB(β A -β B )和B(β B -β B )三种形式。其通过自分泌和旁分泌作用增加垂体FSH的合成和分泌,从而达到促进卵泡发育的作用。激活素依次与两种已知的Ⅱ型激活素受体之一——ActRⅡA或ActRⅡB,以及Ⅰ型受体,激活素受体样激酶4(ALK4)相互作用。FSHβ的表达对激活素的刺激作用极其敏感,其通过促进FSHβ和GnRH受体的转录,与GnRH产生协同作用,故也称为激活素。
卵泡抑素富含半胱氨酸,是一种单体蛋白,不同于激活素和抑制素,在结构上不属于TGF-β家族。在功能上可作激活素的高亲和力结合蛋白,与激活素形成复合物,对FSH有较强的抑制作用,因此又称为卵泡刺激素抑制蛋白(FSP)。垂体的滤泡星形细胞和促性腺激素细胞中卵泡抑制素的合成受到激活素和GnRH的控制,并且会受到性腺类固醇进一步的调节。在女性的月经周期并未发现激活素的变化,然而,在大鼠的动情周期中,垂体中的卵泡抑素的mRNA会发生变化,表明激活素对FSH合成和分泌的影响可能通过卵泡抑素的变化来调节。
综上所述,激活素-抑制素-卵泡抑素系统及BMP在调节FSH的合成和分泌方面与GnRH一起发挥重要作用。此外,该系统也是参与卵泡发育的主要因子。可通过自分泌/旁分泌调节颗粒细胞的增殖和分化,影响膜细胞,调节卵子的成熟及黄体的功能,进而在调节卵巢功能中发挥作用。
低剂量雌激素可以促进促性腺激素的分泌,例如在芳香化酶缺乏症患者、绝经后和切除卵巢的女性中检测到促性腺激素水平升高,是雌激素负反馈调控LH和FSH最直接的证据。有研究表明,在大鼠中切除卵巢后,FSH和LH分泌增加,LHβ、FSHβ和α-mRNA的表达量增加,表达LHβ的细胞数目增加,细胞体积增加,单细胞的总表达量增加。在大鼠中用下丘脑灌注技术或在山羊和猴子中垂体门脉循环插管测量GnRH分泌的研究表明,卵巢切除后GnRH增加,雌激素替代治疗后GnRH随之增加。雌二醇的摄入与大鼠下丘脑组织切片以及GnRH神经元细胞系中GnRH表达量的降低有关。在 ER 敲除的小鼠中的研究表明两种位于下丘脑中的ERα在雌激素负反馈中的重要作用。下丘脑雌激素负反馈效应是通过GnRH上游的神经元介导的,最主要的是位于正中隆起的KNDy神经元。位于视前区的雌激素受体GABA神经元也在介导雌激素对GnRH的负反馈作用中起作用。
女性中雌激素负反馈的机制在绝经后妇女或卵巢切除的女性中得到了广泛的研究,有证据表明,GnRH因性腺类固醇激素反馈而降低,但在绝经后妇女随着年龄的增长而增加。与绝经前妇女相比,利用GnRH拮抗剂最大剂量给药来估测体内的GnRH量,发现在绝经后GnRH水平增高,与绝经后妇女尸检中GnRH mRNA的增加量一致。随着低剂量雌激素的摄入,绝经后妇女的GnRH水平恢复至卵泡期水平,表明在缺乏性腺反馈的条件下,GnRH的增加完全可以归因于雌激素的缺失。此外,Ottowitz等利用神经影像学的研究发现,绝经后妇女短期接触低剂量的雌激素与下丘脑内侧基底代谢活动减少相关。在绝经后妇女的大多数研究中,雌激素给药不会降低GnRH脉冲频率;因此,雌二醇对下丘脑的负反馈作用可能是通过GnRH脉冲振幅的变化来介导的。这些结果与卵巢切除的绵羊和猴子的垂体门脉血液中直接测量GnRH的结果类似,表明GnRH脉冲幅度的下降而不是雌二醇给药的频率介导雌激素对下丘脑的负反馈作用。
尽管上述报道的研究表明雌激素负反馈作用于下丘脑,但是也应考虑作用于垂体的部位。在培养的垂体细胞中,雌二醇会瞬间降低LH对GnRH的反应,且将雌二醇给予下丘脑损伤的猴子接受脉冲GnRH可以降低LH的分泌。ERα和ERβ都存在于促性腺激素细胞上,并且促性腺激素细胞特异性ERα(ESR1)敲除的小鼠已经提供了雌二醇对啮齿动物垂体的直接抑制作用的确凿证据。Shawn等近期从绝经后妇女分离出的下丘脑中的实验已经将从低等生物的研究拓展到人类,证实了生理水平的雌二醇对垂体的促性腺激素分泌有直接影响,且对FSH的影响大于LH。
孕酮对促性腺激素分泌具有重要影响,表现为在下丘脑水平通过减缓脉冲GnRH分泌表现出来。该效应需要雌激素来激发,可能是通过上调下丘脑的孕酮受体而实现的。在接受低剂量雌二醇的绝经后妇女中,使用LH作为GnRH分泌的标志物,加用孕酮可以均一地抑制GnRH脉冲频率,并且应用孕酮可降低GnRH分泌的总量。孕酮有可能是通过直接和间接机制发挥其对GnRH分泌的作用。如前所述,有充分的证据表明,β-内啡肽系统在调节孕酮对GnRH脉冲频率的作用中发挥关键的作用,并且在绵羊的研究中表明孕酮对GnRH的分泌的抑制作用是由强啡肽介导的。
早在20世纪初,就有证据表明一类非甾体性腺因子对垂体具有反馈作用,但直到20世纪80年代才开始分离得到,这类非甾体类性腺因子,包括抑制素A、抑制素B、激活素和卵泡抑素。抑制素B可在男性的血液循环中发现,而抑制素A和抑制素B在育龄期女性的血液和卵泡液中均能检测到。
在卵泡的发生过程中,抑制素A在排卵前达到峰值水平,抑制素A与雌二醇类似,与正常月经周期中的优势卵泡的大小相关,卵泡期抑制素A主要是颗粒细胞的产物;然而,有一些证据表明成熟卵泡中的抑制素A是由间质细胞产生。现在的研究证实抑制素A是在卵泡发育期的早期阶段合成和分泌的。与抑制素A相反,血清中抑制素B的模式表明它主要由小窦卵泡分泌。在自发排卵周期的女性中,抑制素B水平与优势卵泡大小无明显相关性。表明抑制素B与优势卵泡生长无关。Basciani等研究显示卵泡液中的抑制素B水平不随卵泡的大小或成熟度变化且抑制素B的合成是局限在颗粒细胞中的,在间质细胞中缺乏其合成。
1)促性腺激素对抑制素A和抑制素B的调节作用:
有研究显示抑制素B分泌增加,与生理水平的FSH刺激早期卵泡发育有一定的相关性,黄体-卵泡过渡期间FSH升高与抑制素B伴随增加的关系可提示这一结果。在GnRH缺乏的女性中,与每90分钟的早期卵泡期频率相比,每4小时较慢的黄体期频率更换GnRH脉冲不仅导致在黄体-卵泡过渡期间无法正常上升,而且导致卵泡无法生长且抑制素B分泌不增加。该研究表明,在卵泡发育的早期阶段,血清中的抑制素B对生理范围内的FSH刺激变化呈显著的敏感性。
Potorac等在为了检测在卵泡发育的不同阶段对不同的促性腺激素的反应的研究中,使用有效的GnRH激动剂下调内源性GnRH后,实验组女性摄入人重组LH(rhLH)或FSH(rhFSH)。当干预达7天时,单独的rhLH对卵泡生长或激素分泌没有影响,但是每天皮下注射rhFSH导致FSH在正常早期卵泡期水平正常,抑制素B分泌增加,提前于雌二醇和抑制素A。该项研究的其中一种解释为,FSH直接刺激颗粒细胞分泌抑制素B,然而,在该发育阶段用FSH干预还导致一群卵泡募集到卵泡生长池中,并显著刺激颗粒细胞的数量。
为了确定卵泡早期发育过程中抑制素B的增加是直接刺激颗粒细胞分泌还是由颗粒细胞数目增加引起的,利用了卵巢切除手术(非卵巢指征)的标本获得的窦前卵泡和窦卵泡进行研究。这些研究表明抑制素B是由窦前卵泡分泌,不同的是,FSH刺激颗粒细胞分泌抑制素A。总之,这些研究表明,抑制素B由颗粒细胞组成性分泌,并且体内观察到,抑制素B在FSH刺激下的增加是由于颗粒细胞数量的增加而不是FSH直接刺激分泌。在卵泡发育的后期阶段,LH和FSH均刺激优势卵泡中抑制素A和雌二醇的分泌,但对抑制素B没有影响。
2)抑制素A或抑制素B的内分泌负反馈作用的证据:
抑制素亚基在包括肾上腺在内的多种组织中表达,然而循环二聚体抑制素的主要来源之一是性腺,且有证据表明,抑制素在抑制垂体FSH分泌中的主要作用机制是内分泌作用。尽管抑制素A或抑制素B的内分泌作用在正常的月经周期中与FSH的关系并不能定论,但是有几个证据表明抑制素确实在FSH的反馈调节中发挥作用。抑制素最初发现于体外培养的垂体细胞中,因其抑制FSH分泌而被关注。绝经后妇女生理水平的性腺类固醇激素的摄入未能使FSH水平恢复正常,可以充分说明抑制素在女性正常生理状态下调节FSH的分泌作用。许多研究者使用生殖衰老模型来证实,随年龄的增长,FSH水平在LH增加或雌二醇减少之前即增加。一系列研究表明,FSH水平的增加和抑制素B水平的减少与生殖衰老呈负相关。
生殖衰老与女性30岁后的生育率的下降有关,在35岁后女性生育率加速下降。与此同时,卵巢的卵泡池逐渐减小,也是生育率下降的一个潜在原因。在35岁时也会首次观察到卵泡期FSH水平的升高。
在35岁或以上且具有正常排卵周期以及卵泡期FSH水平在正常范围内的女性中,仅在早期卵泡期FSH有小幅度但是显著的增加,整个卵泡期抑制素B水平降低,雌二醇水平在早期卵泡期无差异,但在卵泡中期和晚期卵泡期增加。在黄体期,抑制素B、抑制素A和孕酮在较年老但仍来月经的女性中水平较低,而雌二醇水平维持正常。因此,抑制素B的水平最早减少发生在卵泡加速耗竭的时期,提示围绝经期的到来,表明较低的抑制素B水平反映了随着年龄增长卵泡数目的减少。而在雌二醇水平不改变的情况下,抑制素B的这种变化与高水平的FSH相关,为抑制素B在生殖衰老女性中控制FSH分泌中的内分泌作用提供证据。生殖衰老过程的后期阶段进行的研究已经统一证实FSH的增加与正常甚至更高水平的雌二醇和较低抑制素B相关,并进一步支持抑制素B在控制FSH中的负反馈作用。
虽然生殖衰老的相关研究提供了抑制素B对正常女性FSH分泌的负反馈作用的证据,但是这些研究尚未阐明抑制素A或抑制素B相对于雌二醇在正常生殖周期中动态控制FSH中的作用。其中一个关键问题是抑制素A和抑制素B是否会促进在FSH卵泡中期的下降,这一问题对于女性正常生殖周期的单个卵泡发育至关重要。而且研究无法通过给药或者阻断剂直接确定抑制素的作用。但是,可以通过研究对FSH负反馈的雌激素成分,从而推断出抑制素的生理作用。在黄体-卵泡过渡期通过雌二醇给药维持雌二醇水平的研究可证明,抑制素A不参与黄体期FSH的负反馈控制。另一种方法是阻断雌激素的负反馈,使用他莫昔芬阻断ER可以评估雌二醇和抑制素在FSH的负反馈中的相关性。结果表明早卵泡期雌二醇水平减低导致FSH负反馈作用,如果在GnRH缺乏女性中接受GnRH补充治疗则没有这种效应。然而,FSH并未达到更年期水平,表明抑制素B也是正常月经周期中FSH调节的重要因素,这些研究还认为雌二醇和抑制素A都是黄体期抑制FSH分泌所必需的。
FSH的控制不仅依赖于抑制素和雌二醇,还依赖于激活素/卵泡抑素系统。激活素在卵巢和垂体中起到局部生长和分化因子的作用。在正常的月经周期中,总激活素A水平在月经中期和黄体卵泡转化期最高。然而,卵泡液中激活素A作为卵泡发育的功能指示无明显变化,月经周期中游离激活素没有变化,在卵泡和黄体期之间激活素B明显差异。此外,卵泡液中激活素的潜在内分泌功能要和卵泡抑素结合起来考虑,卵泡抑素在许多组织(包括垂体)中合成。尽管已经在血清中测量激活素,但循环中的激活素与循环中的卵泡抑素的亚型FS315不可逆地结合。迄今为止,尚未在组织内鉴定出可改变卵泡抑素中和作用的机制,因此推断,激活素以自分泌和旁分泌而不是内分泌的方式来调节FSH的分泌。
促性腺激素释放缓解因子(gonadotropin surge attenuating factor,GnSAF)也被称为促性腺激素抑制因子(gonadotropin surge inhibiting factor,GnSIF),是一种可以降低下丘脑中LH分泌的因子。尽管经过了多年的探索,GnRH的分子结构尚未完全解析出来,目前已发布了不同的分子序列,但其中只有一个已经显示出与人类基因组的同源性,其分子链为12.5kDa,与人的血清蛋白羧基末端片段具有同源性,且在体外表达GnSAF的生物活性。最早推测其可能的作用是防止早期LH的激增并导致排卵前卵泡的过早黄体化,然而,现在有动物模型和女性的实验证据表明,GnSAF生物活性与卵泡大小之间存在反比关系,小生长卵泡中的浓度最高,表明其主要作用可能是卵泡发育的早期阶段。
雌激素除了抑制促性腺激素的分泌,也发挥刺激作用,促使在排卵前产生LH峰。关于理解促性腺激素峰的形成机制有两个关键的问题:①雌激素如何对LH分泌产生抑制和刺激作用;②雌激素的正反馈部位是否位于垂体、下丘脑,或两者兼有?雌激素的反馈方向取决于雌二醇的暴露程度及雌二醇给药水平的持续时间。充分的实验证据表明,高水平的雌激素增加了跨物种对GnRH的垂体反应性。
孕激素的主要作用除了减慢GnRH分泌频率之外,其对垂体还具有间接和直接作用,导致LH水平的增加。通过对GnRH缺乏的个体的研究,发现脉冲GnRH给药的频率与LH脉冲振幅呈负相关,使得LH脉冲振幅随前一脉冲的持续时间延长而增加。对接受脉冲GnRH给药(含或不含孕激素)的GnRH缺乏妇女的研究表明,孕激素还通过直接对垂体的作用增加LH脉冲幅度,该作用与脉冲频率的变化无关。
抑制素A在女性排卵前升高,抑制素A可能在垂体水平发挥正反馈的作用。在垂体细胞的培养过程中发现,抑制素A将GnRH受体增加3~6倍,且在绵羊体内实验中也发现了类似的结果。雌二醇和抑制素A对GnRH受体的作用是叠加的,抑制素A对LH阳性细胞百分比与结合GnRH的LH阳性细胞百分比的效应要大于雌二醇的作用;然而,抑制素A对于LH受体下游的正反馈作用不如雌二醇的作用显著。
近年来,一些实验结果表明,kisspetpin除了已经充分研究的下丘脑的作用之外,还可能有垂体作用。 Kiss1 和 Kiss1R 在啮齿类动物的促性腺激素细胞和其他垂体细胞中表达,并且 Kiss1 的表达通过雌激素在ERα受体的直接作用促性腺激素细胞的水平被上调。kisspetpin诱导LβT2细胞中 LHβ 和 FSHβ 基因的转录,并在该细胞中增加GnRHR的表达。在雌性小鼠中刺激素诱导的LH峰期间, Kiss1R 表达增强。可能是通过月经中期对GnRH分泌增加的影响及其对 Kiss1R 的刺激作用。虽然在猴子的垂体前叶中发现了kisspeptin阳性细胞,但是尚未证实其与促性腺激素细胞的共定位;在人类中发现了KISS1R表达于垂体中,但目前尚无法确定在月经中期雌二醇水平较高的情况下,该受体表达的改变是否有助于GnRH对垂体的致敏性。
肾上腺皮质分泌的激素有多种,均属类固醇激素。具有生理活性的仅有:糖皮质激素、盐皮质激素和性激素。其中性激素包括少量雄激素和微量雌、孕激素。在多囊卵巢综合征(PCOS)和不孕症中,LH分泌升高,且通常伴随肾上腺皮质功能的障碍。人肾上腺素可以在束状带和网状结构中表达 LHR 基因,另有间接证据表明LH可能影响肾上腺雄性激素的分泌。病理状况下,PCOS患者LH升高,直接作用于卵泡膜细胞,增强CYP17A1活性,从而导致卵泡膜细胞产生过多的雄激素。另外,雄激素分泌过多,可抑制下丘脑分泌GnRH,对抗雌激素,并使卵巢功能受到抑制而出现闭经及男性化表现。
甲状腺激素是酪氨酸的碘化物,包括甲状腺素(T 4 )和三碘甲腺原氨酸(T 3 )和极少量的逆三碘甲腺原氨酸(rT 3 )。甲状腺激素通过:①直接参与和影响卵巢雌激素的代谢;②FSH和LH的分泌调节卵巢的功能,少量的甲状腺激素促进LH的分泌,适量的甲状腺激素维持垂体与性腺功能的平衡,大量的甲状腺激素则抑制性腺激素的分泌;③降低卵巢对垂体促性腺激素的反应性,对卵巢产生直接的抑制作用。
胰岛素具有广泛的生理功能,促进细胞增殖、分化,并参与蛋白质、脂肪和糖类的合成与代谢。胰岛素在维持正常的卵巢功能具有重要的作用。当胰岛素受体后信号传导途径(PI3K通路和MAPK通路)发生异常时,就可能使胰岛素生理功能发生异常,进而可能导致胰岛素抵抗。在由胰岛素拮抗导致的高胰岛素血症患者中,过量的胰岛素将促使卵巢产生过多雄激素,从而发生雄激素血症,导致月经失调,甚至闭经。
中枢神经系统的神经细胞产生的内源性阿片样物质对女性生殖内分泌系统的调节既有中枢作用(松果体、下丘脑、脑垂体),也有外周作用(卵巢、子宫、胎盘等)。内源性阿片肽对下丘脑-垂体系统及生殖激素(GnRH-GnH)呈抑制性调节作用。性激素只能通过负反馈机制影响下丘脑和垂体的分泌功能,而阿片肽作用于HPO轴却可以受生理和病理不同情况的影响。有研究发现,性激素的反馈机制调节下丘脑视前区阿片肽与μ受体的相互作用,会进一步影响生殖功能。
催产素由下丘脑合成经垂体释放入血液。能够促进乳腺的发育和乳汁分泌,并参与其他生理功能的调节。对生殖活动的调节作用也较为复杂。催产素可从如下四方面影响卵巢的功能:①调控卵巢内LHR的数量,刺激LHR的生成;与LH共同促进黄体的形成并维持孕激素的分泌。②促孕酮的生成,为孕酮的生成提供底物。③降低孕酮的分解过程。④高浓度催产素可抑制颗粒细胞产生孕酮。其中闭经泌乳综合征患者由于无排卵与雌激素水平低落,而血中催产素的浓度可异常增高,临床上表现为闭经、泌乳与不孕。
综上所述,女性卵巢功能主要受HPO轴自上而下的神经内分泌调控,同时也受抑制素-激活素-卵泡抑素系统的调节,从而形成一个闭式环路反馈系统。其生理活动受到大脑皮质神经中枢的影响,如外界因素、精神因素等均可影响月经周期。任何一个环节出现功能异常或障碍均可引起卵巢功能紊乱,导致不孕症的发生。
(沈 薇 马菱蔚)
1.Rodgers RJ, Irving-Rodgers HF, van Wezel IL, et al.Dynamics of the membrana granulosa during expansion of the ovarian follicular antrum. Mol Cell Endocrinol, 2001, 171: 41-48.
2.McConnell NA, Yunus RS, Gross SA, et al.Water permeability of an ovarian antral follicle is predominantly transcellular and mediated by aquaporins. Endocrinology, 2002, 143: 2905-2912.
3.Aittomaki K, Lucena JL, Pakarinen P, et al.Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell, 1995, 82: 959-968.
4.Themmen APN, Huhtaniemi IT.Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev, 2000, 21: 551-583.
5.Danilovich N, Javeshghani D, Xing W, et al.Endocrine alterations and signaling changes associated with declining ovarian function and advanced biological aging in follicle-stimulating hormone receptor haploinsufficient mice. Biol Reprod, 2002, 67: 370-378.
6.Zeleznik AJ, Little-Ihrig L, Ramasawamy S.Administration of insulin-like growth factor I to rhesus monkeys does not augment gonadotropin-stimulated ovarian steroidogenesis. J Clin Endocrinol Metab, 2002, 87: 5722-5729.
7.Johnson MT, Freeman EA, Gardner DK, et al.Oxidative metabolism of pyruvate is required for meiotic maturation of murine oocytes in vivo. Biol Reprod, 2007, 77: 2-8.
8.Kim CH.Androgen supplementation in IVF. Minerva Ginecol, 2013, 65: 497-504.
9.Polyzos NP, Davis SR, Drakopoulos P, et al.Testosterone for poor ovarian responders: lessons from ovarian physiology. Reprod Sci, 2018, 25: 980-982.
10.Pradeep PK, Li X, Peegel H, et al.Dihydrotestosterone inhibits granulosa cell proliferation by decreasing the cyclin D2 mRNA expression and cell cycle arrest at G1 phase. Endocrinology, 2002, 143: 2930-2935.
11.Shoham Z.The clinical therapeutic window for luteinizing hormone in controlled ovarian stimulation. Fertil Steril, 2002, 77: 1170-1177.
12.Zhang FP, Poutanen M, Wilbertz J, et al.Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol, 2001, 15: 172-183.
13.Yariz KO, Walsh T, Uzak A, et al.Inherited mutation of the luteinizing hormone/choriogonadotropin receptor (LHCGR) in empty follicle syndrome. Fertil Steril, 2011, 96: e125-130.
14.Russell DL, Robker RL.Molecular mechanisms of ovulation: co-ordination through the cumulus complex. Hum Reprod Update, 2007, 13: 289-312.
15.Duggavathi R, Volle DH, Mataki C, et al.Liver receptor homolog 1 is essential for ovulation. Genes Dev, 2008, 22: 1871-1876.
16.Duffy DM, Stouffer RL.The ovulatory gonadotrophin surge stimulates cyclooxygenase expression and prostaglandin production by the monkey follicle. Mol Hum Reprod, 2001, 7: 731-739.
17.Norman RJ.Reproductive consequences of COX-2 inhibition. Lancet, 2001, 358: 1287-1288.
18.Pall M, Friden BE, Brannstrom M.Induction of delayed follicular rupture in the human by the selective COX-2 inhibitor rofecoxib: a randomized double-blind study. Hum Reprod, 2001, 16: 1323-1328.
19.Park JY, Su YQ, Ariga M, et al.EGF-like growth factors as mediators of LH action in the ovulatory follicle. Science, 2004, 303: 682-684.
20.Peluffo MC, Murphy MJ, Baughman ST, et al.Systematic analysis of protease gene expression in the rhesus macaque ovulatory follicle: metalloproteinase involvement in follicle rupture. Endocrinology, 2011, 152:3963-3974.
21.Garcia V, Kohen P, Maldonado C, et al.Transient expression of progesterone receptor and cathepsin-l in human granulosa cells during the periovulatory period. Fertil Steril, 2012, 97: 707-713 e1.
22.Gautier J, Norbury C, Lohka M, et al.Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+. Cell, 1988, 54: 433-439.
23.Dunphy WG, Brizuela L, Beach D, et al.The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell, 1988, 54: 423-431.
24.Lee MG, Nurse P.Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature, 1987, 327: 31-35.
25.Mehlmann LM, Saeki Y, Tanaka S, et al.The Gs-linked receptor GPR3 maintains meiotic arrest in mammalian oocytes. Science. 2004, 306: 1947-1950.
26.Kawamura K, Sudo S, Kumagai J, et al.Relaxin research in the postgenomic era. Ann N Y Acad Sci, 2005, 1041: 1-7.
27.Sagata N.What does Mos do in oocytes and somatic cells?Bioessays, 1997, 19: 13-21.
28.Choi T, Fukasawa K, Zhou R, et al.The Mos/mitogen-activated protein kinase (MAPK) pathway regulates the size and degradation of the first polar body in maturing mouse oocytes. Proc Natl Acad Sci U S A,1996, 93: 7032-7035.
29.Vaknin KM, Lazar S, Popliker M, et al.Role of meiosis-activating sterols in rat oocyte maturation: effects of specific inhibitors and changes in the expression of lanosterol 14alpha-demethylase during the preovulatory period. Biol Reprod, 2001, 64: 299-309.
30.Xu Z, Williams CJ, Kopf GS, et al.Maturation-associated increase in IP3 receptor type 1: role in conferring increased IP3 sensitivity and Ca 2+ oscillatory behavior in mouse eggs. Dev Biol, 2003, 254: 163-171.
31.Chen J, Melton C, Suh N, et al.Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the oocyte-to-zygote transition. Genes Dev, 2011, 25: 755-766.
32.Matsuda-Minehata F, Inoue N, Goto Y, et al.The regulation of ovarian granulosa cell death by pro-and antiapoptotic molecules. J Reprod Dev, 2006, 52: 695-705.
33.McRae RS, Johnston HM, Mihm M, et al.Changes in mouse granulosa cell gene expression during early luteinization. Endocrinology, 2005, 146: 309-317.
34.Woad KJ, Robinson RS.Luteal angiogenesis and its control. Theriogenology, 2016, 86: 221-228.
35.Sasano H, Suzuki T.Localization of steroidogenesis and steroid receptors in human corpus luteum. Classification of human corpus luteum (CL) into estrogen-producing degenerating CL, and nonsteroid-producing degenerating CL. Semin Reprod Endocrinol, 1997, 15: 345-351.
36.Devoto L, Fuentes A, Kohen P, et al.The human corpus luteum: life cycle and function in natural cycles. Fertil Steril, 2009, 92: 1067-1079.
37.Stouffer RL.Progesterone as a mediator of gonadotrophin action in the corpus luteum: beyond steroidogenesis. Hum Reprod Update, 2003, 9: 99-117.
38.Duncan WC.The human corpus luteum: remodelling during luteolysis and maternal recognition of pregnancy. Rev Reprod, 2000, 5: 12-17.
39.Kohen P, Castro O, Palomino A, et al.The steroidogenic response and corpus luteum expression of the steroidogenic acute regulatory protein after human chorionic gonadotropin administration at different times in the human luteal phase. J Clin Endocrinol Metab, 2003, 88: 3421-3430.
40.Bishop CV, Bogan RL, Hennebold JD, et al.Analysis of microarray data from the macaque corpus luteum, the search for common themes in primate luteal regression. Mol Hum Reprod, 2011, 17: 143-151.
41.Aboelenain M, Kawahara M, Balboula AZ, et al.Status of autophagy, lysosome activity and apoptosis during corpus luteum regression in cattle. J Reprod Dev, 2015, 61: 229-236.
42.Webb R, Woad KJ, Armstrong DG.Corpus luteum (CL) function: local control mechanisms. Domest Anim Endocrinol, 2002, 23: 277-285.
43.Davis JS, Rueda BR.The corpus luteum: an ovarian structure with maternal instincts and suicidal tendencies. Front Biosci, 2002, 7: d1949-1978.
44.Myers M, Lamont MC, van den Driesche S, et al.Role of luteal glucocorticoid metabolism during maternal recognition of pregnancy in women. Endocrinology, 2007, 148: 5769-5779.
45.Marchais-Oberwinkler S, Henn C, Moller G, et al.17beta-Hydroxysteroid dehydrogenases (17beta-HSDs)as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. J Steroid Biochem Mol Biol, 2011, 125: 66-82.
46.Miller WL, Auchus RJ.The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev, 2011, 32: 81-151.
47.Miller WL.Steroidogenesis: unanswered questions. Trends Endocrinol Metab, 2017, 28: 771-793.
48.Rosenfeld CS, Cooke PS.Endocrine disruption through membrane estrogen receptors and novel pathways leading to rapid toxicological and epigenetic effects. J Steroid Biochem Mol Biol, 2019, 187: 106-117.
49.Namwanje M, Brown CW.Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol, 2016, 8: a021881
50.Sonigo C, Beau I, Binart N, et al.Anti-Müllerian hormone in fertility preservation: clinical and therapeutic applications. Clin Med Insights Reprod Health, 2019, 13: 1179558119854755.
51.Victoria M, Labrosse J, Krief F, et al.Anti-Müllerian hormone: more than a biomarker of female reproductive function. J Gynecol Obstet Hum Reprod, 2019, 48: 19-24.
52.Zhang M, Su YQ, Sugiura K, et al.Granulosa cell ligand NPPC and its receptor NPR2 maintain meiotic arrest in mouse oocytes. Science, 2010, 330: 366-369.
53.Zhang M, Su YQ, Sugiura K, et al.Estradiol promotes and maintains cumulus cell expression of natriuretic peptide receptor 2 (NPR2) and meiotic arrest in mouse oocytes in vitro. Endocrinology, 2011, 152: 4377-4385.
54.Limonta P, Marelli MM, Moretti R, et al.GnRH in the human female reproductive axis. Vitam Horm, 2018, 107: 27-66.
55.Ivell R, Anand-Ivell R.Insulin-like peptide 3 (INSL3) is a major regulator of female reproductive physiology. Hum Reprod Update, 2018, 24: 639-651.
56.Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, et al.Arcuate kisspeptin/neurokinin B/dynorphin (KNDy) neurons mediate the estrogen suppression of gonadotropin secretion and body weight. Endocrinology, 2012, 153: 2800-2812.
57.Lehman MN, Hileman SM, Goodman RL.Neuroanatomy of the kisspeptin signaling system in mammals: comparative and developmental aspects. Adv Exp Med Biol. 2013; 784: 27-62
58.Limonta P, Marelli MM, Moretti R, et al.GnRH in the human female reproductive axis. Vitam Horm. 2018; 107: 27-66.
59.Stamou M, Cox K, Crowley WF.Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the“-omics”era. Endocrine reviews, 2015, 2016: 4-22.
60.Goodman RL, Lehman MN, Smith JT, et al.Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology, 2007, 148: 5752-5760.
61.de Roux N, Genin E, Carel JC, et al.Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proceedings of the National Academy of Sciences, 2003, 100: 10972-10976.
62.Teles MG, Bianco SD, Brito VN, et al.A GPR54-activating mutation in a patient with central precocious puberty. New England Journal of Medicine, 2008, 358: 709-715.
63.Rønnekleiv OK, Kelly MJ.Kisspeptin excitation of GnRH neurons. Kisspeptin Signaling in Reproductive Biology: Springer, 2013: 113-131.
64.Lehman MN, Coolen LM, Goodman RL.Minireview: kisspeptin/neurokinin B/dynorphin (KNDy)cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology, 2010, 151: 3479-3489.
65.George J, Hendrikse M, Veldhuis JD, et al.Effect of gonadotropin‐inhibitory hormone on luteinizing hormone secretion in humans. Clinical endocrinology, 2017, 86: 731-738.
66.Bilezikjian LM, Justice NJ, Blackler AN, et al.Cell-type specific modulation of pituitary cells by activin, inhibin and follistatin. Molecular and cellular endocrinology, 2012, 359: 43-52.
67.Ottowitz WE, Dougherty DD, Fischman AJ, et al.[ 18 F] 2-fluoro-2-deoxy-d-glucose positron emission tomography demonstration of estrogen negative and positive feedback on luteinizing hormone secretion in women.The Journal of Clinical Endocrinology & Metabolism, 2008, 93: 3208-3214.
68.Shaw N, Histed S, Srouji S, et al.Estrogen negative feedback on gonadotropin secretion: evidence for a direct pituitary effect in women. The Journal of Clinical Endocrinology & Metabolism, 2010, 95: 1955-1961.
69.Basciani S, Watanabe M, Mariani S, et al.Hypogonadism in a patient with two novel mutations of the luteinizing hormone β-subunit gene expressed in a compound heterozygous form. The Journal of Clinical Endocrinology & Metabolism, 2012, 97: 3031-3038.
70.Nagirnaja L, Venclovas
 , Rull K, et al.Structural and functional analysis of rare missense mutations in human chorionic gonadotrophin β-subunit. Molecular Human Reproduction, 2012, 18: 379-390.
, Rull K, et al.Structural and functional analysis of rare missense mutations in human chorionic gonadotrophin β-subunit. Molecular Human Reproduction, 2012, 18: 379-390.
71.Potorac I, Rivero-Muller A, Trehan A, et al.A vital region for human glycoprotein hormone trafficking revealed by an LHB mutation. Journal of Endocrinology, 2016, 231: 197-207.
72.Şimşek E, Montenegro LR, Binay C, et al. Clinical and hormonal features of a male adolescent with congenital isolated follicle-stimulating hormone deficiency. Hormone Research in Paediatrics, 2016, 85: 207-212.
73.Messinis IE, Messini CI, Anifandis G, et al.Gonadotropin surge-attenuating factor: a nonsteroidal ovarian hormone controlling GnRH-induced LH secretion in the normal menstrual cycle. Vitamins and hormones: Elsevier, 2018: 263-286.
74.Gahete MD, Vázquez-Borrego MC, Martínez-Fuentes AJ, et al.Role of the Kiss1/Kiss1r system in the regulation of pituitary cell function. Molecular and Cellular Endocrinology, 2016, 438: 100-106.
卵巢在女性的生殖寿命中充当启动和维持的积极角色,通过周期性排卵和分泌激素,承担着女性的生育功能,并维持女性机体内分泌环境的稳定。
卵巢是一个动态变化的器官,从胚胎期到绝经过渡期不断发生着显著变化。人胚胎约20周时的始基卵泡数目最多,37岁左右开始卵泡耗竭加速,到最后一次月经时卵巢中所剩的卵泡极少。在从胚胎形成到逐渐老去这一渐进的过程中,卵巢组织的结构和功能都会经历一系列的变化,也是其成长、成熟、逐渐衰老或老化的过程。
卵巢的生命周期包括卵巢的发生、成长、成熟和卵巢功能的衰退及衰竭五个时期。那么,什么是卵巢衰老呢?卵巢衰老的概念在前面章节已提及,主要是指年龄相关的女性卵巢储备/功能逐渐衰退直至衰竭的过程,受遗传、环境、社会心理、生活方式等多因素影响,以卵泡数量和卵子质量下降为基础,最终表现为绝育乃至绝经,并影响全身多个系统和器官,导致相关疾病和综合征的发生、发展。
卵巢衰老与年龄显著相关,多呈年龄依赖性,年龄所致累积性损伤是卵巢衰老的主要影响因素。而卵巢衰老不仅包括了年龄相关的自然衰老,还包括了遗传、环境、行为等多种因素导致的卵巢储备和功能下降,如POI、DOR和POR等。各种因素可通过直接或间接途径损害卵泡数量和质量,导致卵巢储备和功能下降,加速卵巢衰老进程。不同的影响因素在机制方面可能相通。卵巢衰老涉及DNA损伤、表观遗传、端粒缩短、端粒酶活性下降、自由基损伤及线粒体功能下降等机制。目前,卵巢衰老患者病因和机制仍不明确,有待进一步探究。
卵巢衰老以功能为核心,表现为生育能力下降、激素内分泌紊乱以及月经异常,是一个类似于程序化的事件。随着年龄增长,女性卵巢储备和功能逐渐下降。女性在20~30岁达到生育高峰,生育能力平均在30岁开始逐月下降,37.5岁后卵泡数目下降的速度翻倍,呈现“折棍”现象。卵泡作为卵巢的主要内分泌和生殖单元,参与并在一定程度上决定了卵巢的生殖潜能和生殖寿命。卵子质量衰退导致女性生育力和生殖质量下降,是关系到自身及下一代健康的重要问题。
卵巢衰老的解剖学及细胞学基础是卵泡数量和质量下降。随着年龄的增长,除卵母细胞数目持续减少、卵子质量下降以及卵母细胞所在皮质的厚度相应变薄之外,卵巢体积亦不断缩小。卵巢表面上皮发生变化,乳头和隐窝较少见,表面上皮细胞微绒毛较少且较短,凋亡和坏死细胞数量逐渐增加。随着卵巢功能逐渐衰退,卵泡不能发育成熟及排卵,会伴发阴道不规则出血、围绝经期症状及不孕等临床较为常见的合并症。最终由于卵巢内卵泡自然耗竭,对垂体促性腺激素丧失反应,导致卵巢功能衰竭,直至绝经。绝经后功能已衰竭的卵巢,重量不到10g,呈现表面无光泽、有皱纹的外观。从形态学上看,衰老卵巢的主要变化是卵巢体积缩小,间质水肿增多,结缔组织堆积。绝经后5年内,卵巢中仍可看到几个始基卵泡和正在经历成熟和闭锁的卵泡;血管网缩小,血管管腔缩窄,血管壁增厚或硬化,其特征性表现为多普勒超声下看到卵巢基质血流减少。绝经后早期卵巢虽然停止分泌雌激素,但其间质仍能分泌少量雄激素,此期由雄激素在外周转化而来的雌酮成为循环中的主要雌激素。
卵巢衰老最终表现为绝育乃至绝经,以绝经为标志事件。正常健康女性自然受孕能力平均在41岁丧失。中国女性平均绝经年龄约在52岁。女性40岁开始即将经历绝经过渡期,这亦是从生育期到非生育期的过渡时期。机体从临床特征、内分泌、生物学方面开始出现趋向绝经的变化直至最后一次月经的终止,这是卵巢衰老的重要进程,这一过程可能经历数年或10余年,但直到现在,我们对这一时期的变化仍知之甚少。2001年召开的生殖衰老分期(the stages of reproductive aging workshop,STRAW)会议通过评估与生殖衰老相关的月经周期性表现,内分泌学症状、生育力以及卵巢影像学改变,进行了STRAW分期,10年后STRAW对该分期系统更新,提出了STRAW+10分期系统以便于对女性生育能力进行综合评估,有助于做出临床判断并及时给予相应的个体化干预。
卵巢衰老对机体危害极大。绝经后,卵巢功能丧失,机体内分泌失衡,导致或加剧多器官、多系统功能障碍。妇女60岁后进入老年期,此期机体所有内分泌功能普遍低落,卵巢功能已经完全衰竭,主要表现为雌激素水平低落,不足以维持女性第二性征,生殖器官亦逐渐萎缩,除卵巢缩小变硬以外,子宫及子宫颈萎缩;阴道逐渐缩小,穹窿变窄,黏膜变薄、无弹性;阴唇皮下脂肪减少,阴道上皮萎缩,糖原消失,分泌物减少,呈碱性,容易感染导致老年性阴道炎。绝经后期与雌激素减少有关的一系列内分泌代谢改变会引起其他多系统的功能紊乱,如血管舒缩症状,泌尿生殖系统、心血管系统、骨关节及神经系统的症状,严重影响了妇女的生活质量。其中,骨代谢异常引起骨质疏松,易发生骨折。另外,心血管疾病、老年性痴呆、肥胖、肿瘤、糖尿病等的发生率都随着绝经而增加。因此,卵巢衰老被喻为女性机体衰老的起搏器,是多个器官衰老的始动因素。卵巢衰老带来的一系列困扰严重影响家庭幸福指数以及女性健康寿命。重视卵巢衰老伴随的激素水平相关变化,通过一些方式来逆转或缓解这些改变,可显著提高此年龄段女性的生活质量。
随着衰老逐渐被认为是机体的一种病理过程,我们亦提出这样一个观点,卵巢衰老是一个受多因素、多环节影响的长期性、累积性、复杂性的病理过程。这是由于卵巢的衰老不仅危害生殖系统本身,更会加速多器官、多系统功能障碍。卵巢衰老对机体各系统、各器官的危害将在后续章节进行详细阐述。鉴于这些危害的隐匿性、严重性和长期性,卵巢的衰老应该受到足够重视。针对其影响因素和病因的探索、致病机制的挖掘,早期评估和预警体系的建立以及防治策略的前沿探索,应该被重点关注。
(张金金 郝 星)
1.Fédération C, Schwartz D, Mayaux MJNEJoM.Female fecundity as a function of age: results of artificial insemination in 2193 nulliparous women with azoospermic husbands. Federation CECOS. N Engl J Med, 1982, 306: 404-406.
2.Faddy M, Gosden R, Gougeon A, et al.Accelerated disappearance of ovarian follicles in mid-life: implications for forecasting menopause. Hum Reprod, 1992, 7: 1342-1346.
3.Sun X, Luo M, Ma M, et al.Ovarian aging: an ongoing prospective community-based cohort study in middleaged Chinese women. Climacteric, 2018, 21: 404-410.
4.Nelson HD.Menopause. Lancet, 2008, 371: 760-770.
5.Couzin-Frankel J.Reproductive Biology. Faulty DNA repair linked to ovarian aging in mice and humans. Scie nce, 2013, 339: 749.