
下载掌阅APP,畅读海量书库
立即打开




|
1.3 原料气的净化 |

|
无论用何种方法生产出的原料气,都含有一定数量的二氧化碳、一氧化碳、硫化物等对合成反应不利的成分,在进入合成塔之前都必须除去。
 1.3.1 原料气的脱硫
1.3.1 原料气的脱硫
原料气中的硫化物主要是硫化氢,其次是有机硫。它们的存在能够使各种催化剂中毒,且腐蚀管道设备,所以在进一步加工之前,必须先进行脱硫。在天然气为原料的合成氨工艺中,是先用干法脱硫除去天然气中的硫,再进行转化反应的。
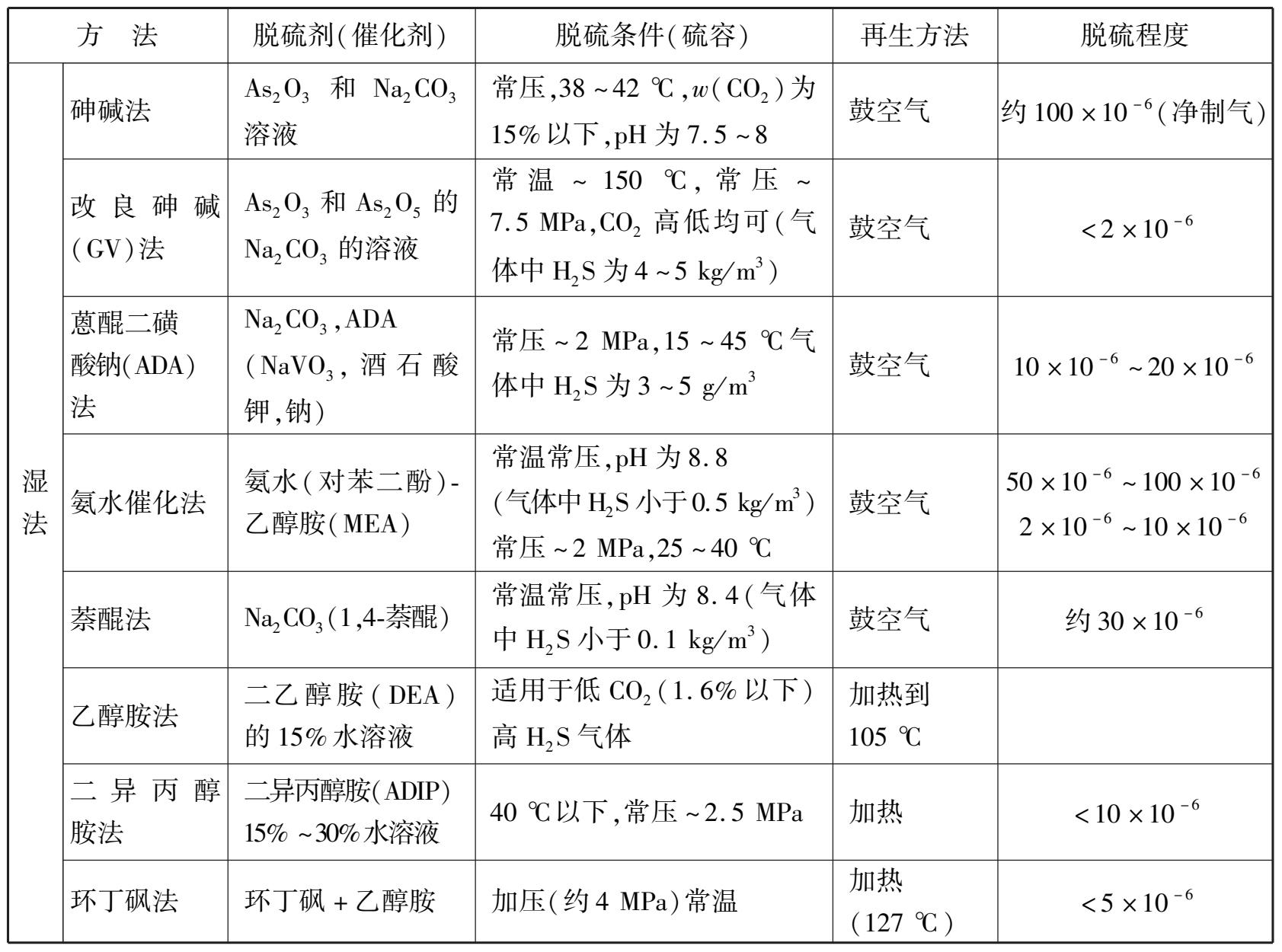
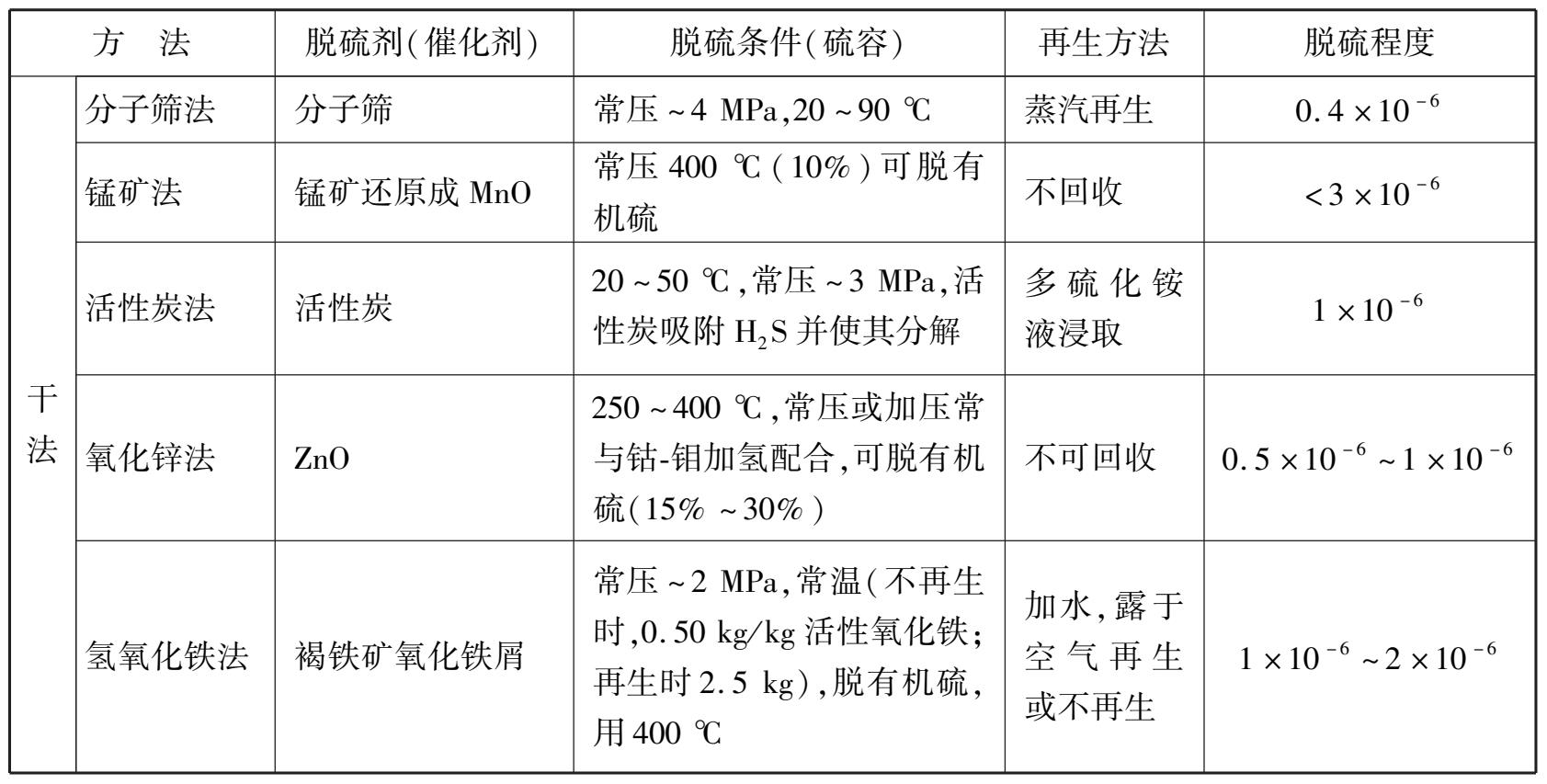
随着科学技术的发展,脱硫的方法日益增多,但根据脱硫剂的物理形态,可分为干法脱硫和湿法脱硫两种,而干法脱硫和湿法脱硫又可分为若干类,如表 1.3 所示。
表 1.3 合成氨原料气脱硫的方法概况
续表
1)干法脱硫
干法脱硫是用固体吸收剂吸收原料气中的硫化物,一般只有当原料气中硫化氢质量浓度不高,标准状态下在 3~5 g/m 3 才适用。常用的干法脱硫有氧化锌脱硫法、钴-钼加氢脱硫法等。
(1)氧化锌脱硫法
以氧化锌为脱硫剂的干法脱硫,是近代合成氨厂广泛采用的精细脱硫的方法,可脱除无机硫和有机硫,脱硫反应为:
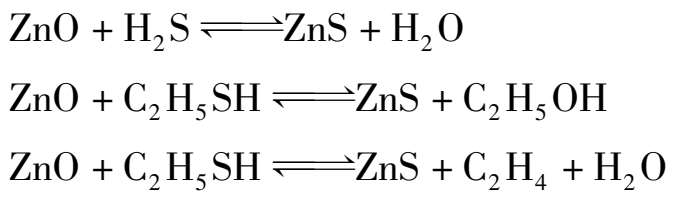
有氢存在时,有些有机硫化物先转化成硫化氢,再被氧化锌吸收,其反应为:
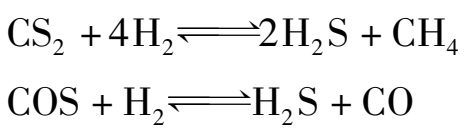
但氧化锌对噻吩(C 4 H 4 S)的转化能力很差。
氧化锌与硫化氢反应的平衡常数
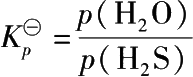
不同温度下的平衡常数为:

由平衡常数可知,在通常情况下脱硫相当完全。有机硫的脱除可认为是氧化锌对某些有机硫化合物的热分解有催化作用,使之分解为碳氢化合物和硫化氢,硫化氢再被吸收。多数有机硫化物在 400℃以下就发生热分解。氧化锌工业脱硫的温度在 200~450℃范围内选择,脱无机硫温度控制在 200℃左右,脱有机硫温度则在 350~450℃范围内。
工业上使用的氧化锌脱硫剂都做成与催化剂一样的多孔结构,脱硫的反应是在氧化锌的微孔内表面上进行的。除了温度、空速等操作条件影响脱硫效率外,氧化锌颗粒的大小、形状和内部孔结构,也影响脱硫效率。颗粒的孔容积越大,内表面越发达,脱硫的效果就越好。
氧化锌脱硫用硫容(每千克氧化锌脱硫剂能吸收H 2 S的千克数)来表示其脱硫性能的好坏。硫容值越大,则氧化锌脱硫的本领就越强。通常氧化锌脱硫的硫容平均为 0.15~0.20 kg/kg,最高达 0.3 kg/kg。硫容与温度的关系如图 1.13 所示。氧化锌脱硫后,出口气体的含硫量小于 1 ×10 -6 。使用过的氧化锌不可再生,因此适用于脱微量硫。当原料气中硫含量较高时,氧化锌脱硫法常与湿法脱硫或其他干法脱硫(如活性炭脱硫)配合使用。
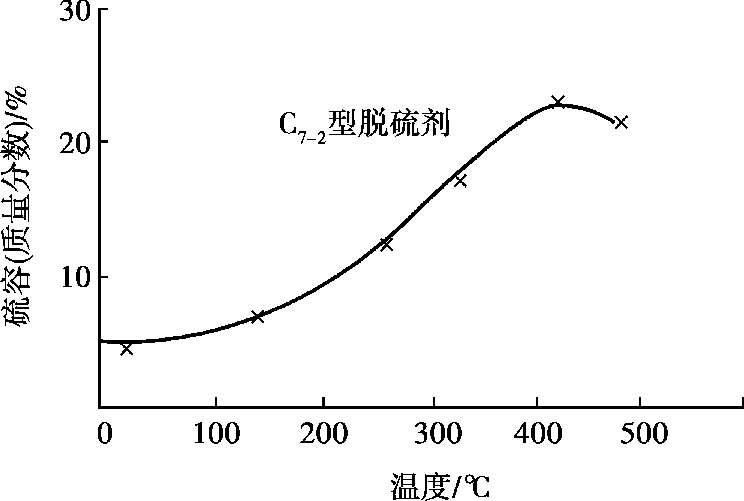
图 1.13 氧化锌脱硫剂的硫容量(空速为 700 h -1 ,进口H 2 S为 5 × 10 -5 ,出口H 2 S < 1 × 10 -6 )
(2)钴-钼加氢脱硫法
钴-钼加氢脱硫法是脱除含氢原料中有机硫十分有效的预处理措施。钴-钼加氢催化剂几乎可使天然气、石脑油原料中的有机硫全部转化成硫化氢,再用氧化锌吸收可把总硫脱除到2.00× 10 -8 以下。在有机硫转化的同时,也能使烯烃加氢转变为饱和的烷烃,从而减少下一工序蒸汽转化催化剂析碳的可能性。
钴-钼加氢催化剂是以氧化铝为载体,由氧化钴和氧化钼所组成。钴-钼催化剂要经硫化后才能呈现活性。硫化后活性组分主要是MoS 2 ,其次是Co 9 S 8 。通常认为MoS 2 提供催化活性,而Co 9 S8主要是保持MoS 2 具有活性的微晶结构,以阻止产生MoS 2 活性衰退的微晶集聚过程。使用过程中,当催化剂表面积累胶质或结炭而导致催化活性下降时,可以通蒸汽再生,或通入含少量空气的氮(氧的体积分数为 1%~1.5%),在 500~550℃的温度下将积炭燃烧而再生。
钴-钼加氢催化剂上,有机硫发生加氢的分解反应为:
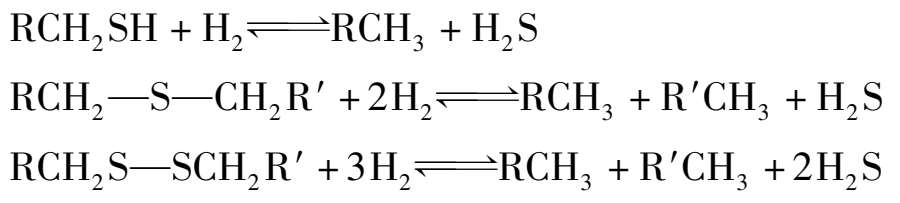
有机硫加氢分解反应是放热反应,平衡常数值随温度的升高而降低。工业生产上,操作温度在 340~400℃范围内,平衡常数值还很大。在此条件下,有机硫的加氢分解实际上是完全的。
钴-钼加氢的工艺条件根据原料烃性质、净化度要求以及催化剂的型号来决定。操作温度为 340~400℃,操作压力则按不同型号催化剂而异。加氢所需的氢量一般维持反应后气体中有 5%~6%的氢气;入口空间速度气态烃为 500~1 500 h -1 ,液态烃为 0.5~6 h -1 。
2)湿法脱硫
干法脱硫的优点是既能脱除有机硫,又能脱除无机硫,而且可以把硫脱至极精细的程度(1× 10 -6 以下)。其缺点是脱硫剂或不能再生,或再生非常困难;而且干法脱硫设备庞大,占地很多。因此不适用于脱除大量无机硫,只有天然气、油田气、炼厂气等含硫较低时才采用干法脱硫。
当需要脱除大量无机硫时,采用湿法脱硫有明显的优点。首先是脱硫剂是便于输送的液体物料;其次脱硫剂可以再生并能回收富有价值的化工原料硫磺,从而构成一个连续脱硫的循环系统。因此当原料气中含硫量较高时,根据工艺要求采用湿法一次脱硫,或湿法粗脱串联干法精脱,以达到工艺上和经济上都合理的目的。
湿法脱硫方法很多,根据脱硫过程的特点可分为化学吸收法、物理吸收法和化学-物理综合吸收法 3 类。化学吸收法是以弱碱性吸收剂吸收原料气中的硫化氢,吸收液(富液)在温度升高和压力降低时分解而释放出硫化氢,解吸后的吸收液(贫液)循环使用。这类方法有乙醇胺法、二异丙醇胺法等。物理吸收法是用熔剂选择性地溶解原料气中的硫化氢,吸收液在压力降低时释放出硫化氢,溶剂可再循环利用,如聚乙二醇二甲醚法等。化学-物理综合吸收法就是将化学物理两种吸收法结合起来,如环丁砜法等。
改良的蒽醌二磺酸钠(Anthraquinone Disulphonic Acid,简写ADA)法为化学吸收,又称改良ADA法,在湿法脱硫中运用最为普遍。该法是在早期ADA法溶液中添加适量的偏钒酸钠作载氧剂、酒石酸钠及少量三氯化铁和乙二胺四乙酸(EDTA)作活化剂,脱硫效果和经济效益大大改善。
改良的ADA脱硫法,在脱硫塔中用pH为8.5~9.2 的稀碱溶液吸收硫化氢并生成硫氢化物:
Na 2 CO 3 +H 2 S═NaHS+NaHCO 3
液相中的硫氢化物进一步与偏钒酸钠反应,生成还原性焦性偏钒酸盐,并析出元素硫。
2NaHS+4NaVO 3 +H 2 O ═Na 2 V 4 O 9 +4NaOH+2S
还原性焦性偏钒酸钠接着与氧化态ADA反应,生成还原态的ADA和偏钒酸盐。
Na 2 V 4 O 9 +2ADA(氧化态)+2NaOH+H 2 O ═4NaVO 3 +2ADA(还原态)
还原态的ADA被空气中的氧氧化成氧化态的ADA,其后溶液循环使用。
2ADA(还原态)+O 2 ═2ADA(氧化态)+H 2 O
当气体中含有二氧化碳、氰化氢和氧时,还会与碱发生副反应,生成NaCNS和Na 2 SO 4 等,其反应如下:
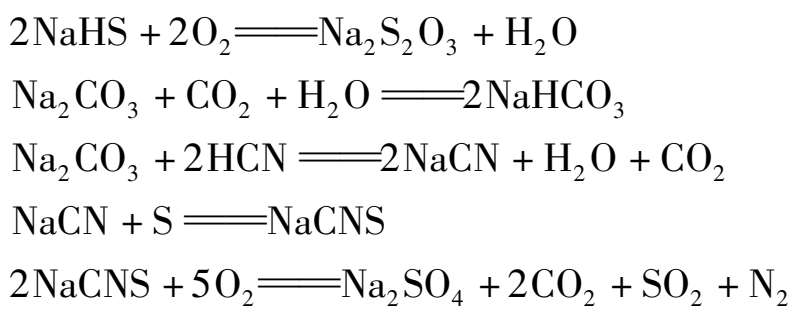
所以,一定要防止硫以氰化钠形式进入再生塔,以免影响ADA的再生。生成NaCNS和Na 2 SO 4 也消耗了碱,对循环脱硫过程不利。因此当溶液中NaCNS和Na 2 SO 4 积累到一定程度后,必须引出一部分溶液,补充相应数量的新鲜溶液,以保持正常生产。
改良ADA法的主要操作条件有:
①溶液的pH值:溶液的pH值对气体吸收程度有很大影响,如图 1.14 所示。提高溶液的碱度,气体中的硫化氢的吸收程度也提高。当pH达到 8.8 时,吸收已基本完全;pH值再提高,吸收液中硫化合物转化成硫代硫酸钠的量增大。吸收液是循环利用的,硫代硫酸钠的积累,降低了碳酸钠和偏钒酸钠的溶解度,也影响硫的回收。通常pH值以 8.5~9.2 为宜。
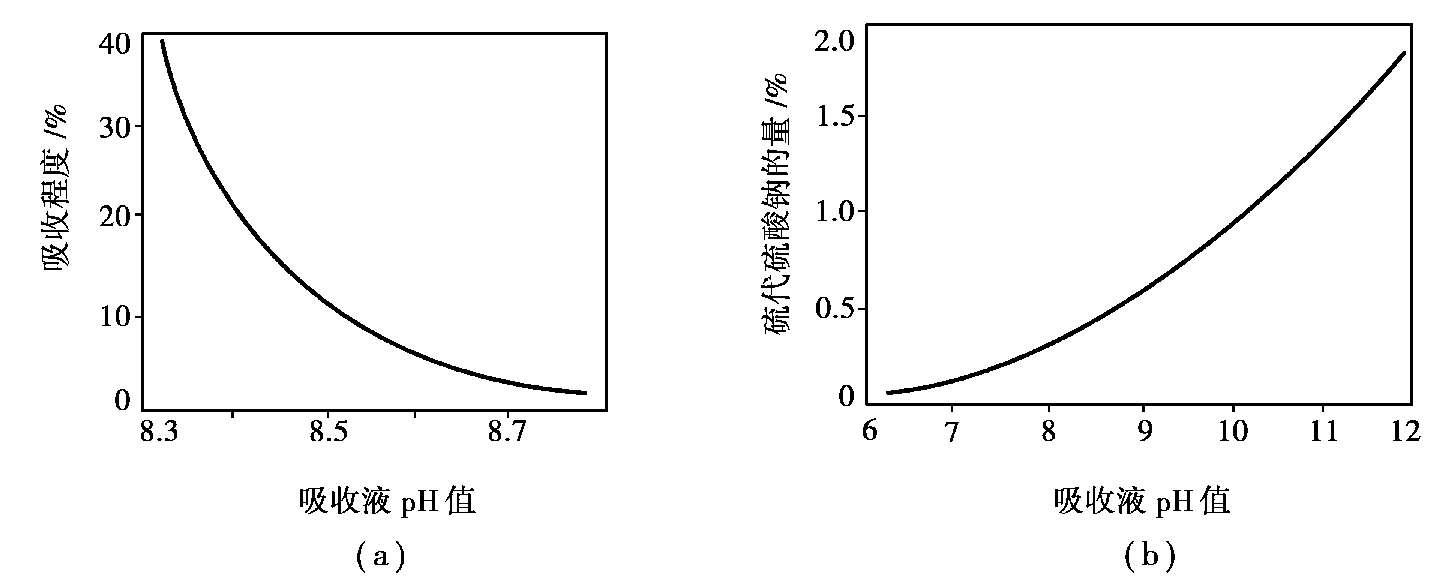
图 1.14 脱硫时pH值的影响
(a)pH值对吸收程度的影响;(b)pH值对生成硫代硫酸钠的影响
②钒酸盐含量的影响:溶液中偏钒酸钠与硫氰化物的反应是相当快的,一般 5 min即可完成。为防止硫化氢局部过量生成“钒-氧-硫”的黑色沉淀,应使偏钒酸钠量比理论值稍大,且使溶液中ADA含量必须等于或大于偏钒酸含量的 1.69 倍,工业上实际采用 2 倍左右。
③温度:温度对脱硫的影响如图 1.15 所示。过高的温度促使大量的硫代硫酸钠副反应发生,低的吸收温度则使吸收和再生速度变慢,生成的单质硫粒径也只有 0.5~5μm而不易分离。生产中常维持溶液的吸收温度为 40~50℃。
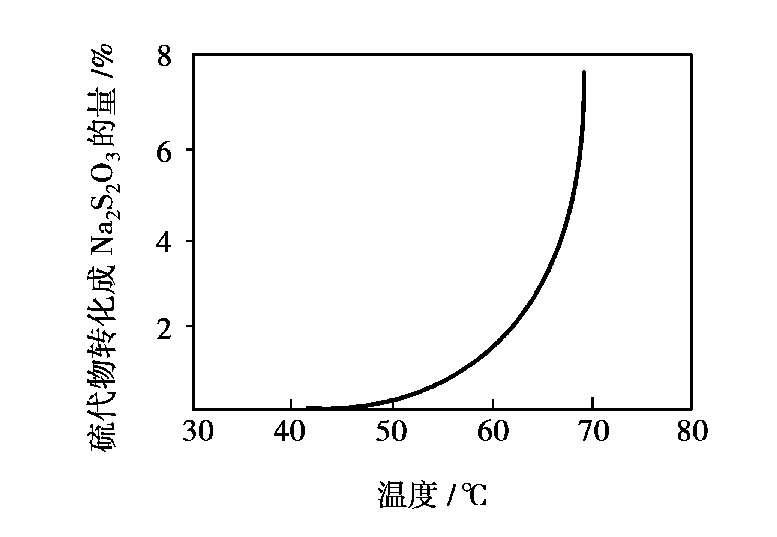
图 1.15 脱硫时温度的影响
④压力:用改良ADA法脱硫时,一般在加压和常压下进行。吸收压力的大小取决于原料气本身的压力或脱硫工序在合成氨流程中的部位。加压可提高设备的生产强度,减少设备容积,提高气体的净化度。但吸收压力过高,氧在溶液中的溶解度增大,会加快副反应的速度。
⑤再生空气用量和再生时间:再生空气量除满足氧化还原态ADA的需要外,还应使硫呈泡沫状悬浮在溶液表面上,以便溢流回收。标准状态下一般再生塔空气量控制在 80~120 m 3 /(m 2 ·h)为宜。溶液在再生塔中停留时间一般为 30~40 min,不宜太长,否则会大大增加再生器容积。
改良ADA法的工艺流程如图 1.16 所示。
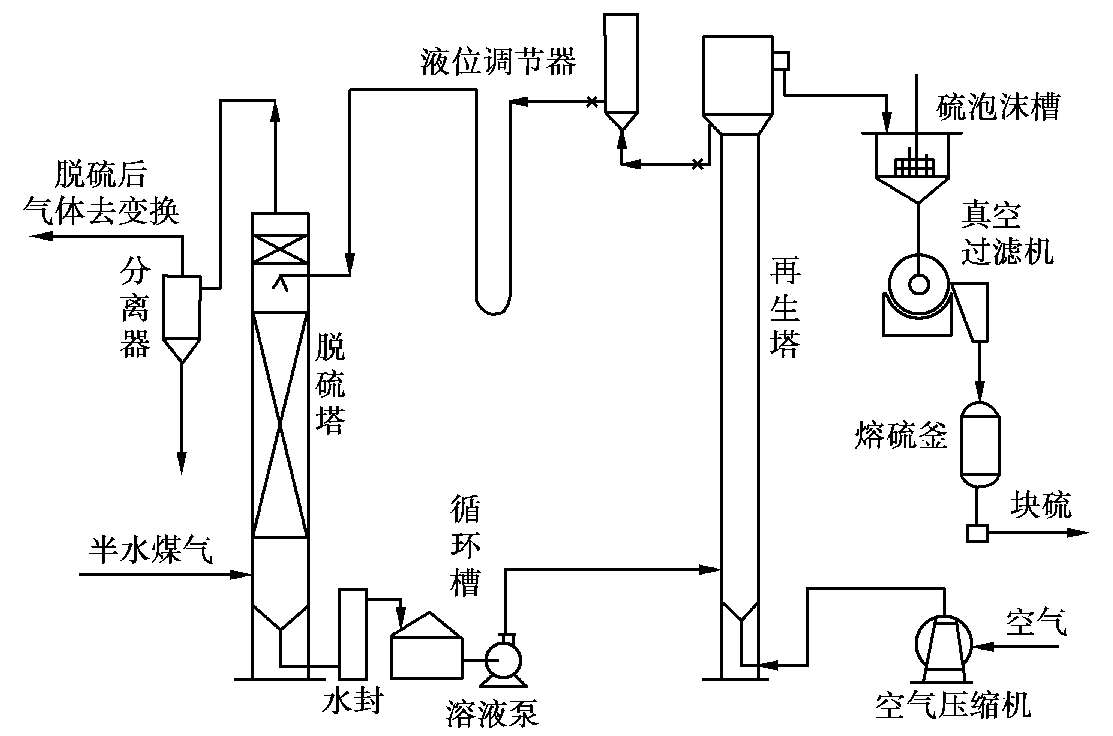
图 1.16 改良ADA法的工艺流程简图
含有硫化氢的原料气从脱硫塔底部进入,与塔顶喷淋下的溶液逆流接触,气体中的硫化氢被溶液吸收而脱除,从脱硫塔顶出来送往下一工序。吸收硫化氢后的溶液从脱硫塔底部引出,经循环槽用泵打入再生塔进行再生,空气由塔顶排出,析出的硫泡沫由塔顶的扩大部分上部溢流入硫泡沫槽,用真空过滤机分离出硫磺,滤液返回循环槽(图中未示出)。氧化后的溶液再由再生塔顶部扩大部分的下部出口引出,经液位调节器进入脱硫塔循环使用。
1.3.2 一氧化碳变换
用不同燃料制得的合成气,均含有一定量的一氧化碳。一般固体燃料气化制得的水煤气中一氧化碳的体积分数为 35%~37%,半水煤气中一氧化碳的体积分数为 25%~34%,天然气蒸汽转化制得的转化气中一氧化碳的体积分数较低,一般为 12%~14%。一氧化碳是合成甲醇的直接原料,但不是合成氨生产所需要的直接原料,而且在一定条件下还会与合成氨的铁系催化剂发生反应,导致催化剂失活。因此,在原料气进入合成塔之前,必须将一氧化碳清除。清除一氧化碳分两步进行,首先进行变换反应:
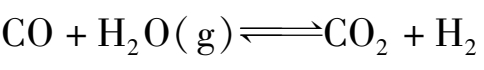
这样,既能把大部分一氧化碳变为易于清除的二氧化碳,而且又制得了等摩尔量的氢,而所消耗的只是廉价的水蒸气。因此,一氧化碳变换既是原料气的净化过程,又是原料气制造的继续。少量残余的一氧化碳再通过其他净化法如甲烷化加以脱除。
工业中,一氧化碳变换均在有催化剂的条件下进行。20 世纪 60 年代以前,主要采用以Fe 2 O 3 为主体的催化剂,使用温度为 350~550℃。由于一氧化碳变换反应为可逆放热反应,受操作温度的限制,气体经变换后仍有 3%左右的一氧化碳。20 世纪 60 年代以来,由于脱硫技术的发展,使得在更低温度下使用活性高而抗毒性差的CuO催化剂成为可能,CuO催化剂的操作温度在 200~280℃,该温度下残余一氧化碳可降至 0.3%左右。
为了区别上述两种温度范围的变换过程,将前者称为中温变换(或高温变换),而后者称为低温变换。所用催化剂分别称为中变(或高变)催化剂及低变催化剂。
1)变换反应热力学
一氧化碳和水蒸气的变换反应为放热反应,平衡常数随温度升高而降低。平衡常数可用式(1.2.4)计算。
现以 1 mol原料气为基准,用 y a , y b , y c 和 y d 分别代表初始气体中的CO,H 2 O,CO 2 和H 2 的摩尔分数, x 为变换反应的平衡转化率(或变换率)。
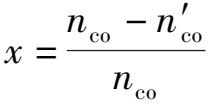
式中 n co ——进口CO摩尔量;
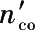 ——出口CO摩尔量。
——出口CO摩尔量。
通常生产中测量的是不含水蒸气的气体组成,称为干基组成。如果分析该变换前后CO的干基组分分别为 y 和 y′ ,则 1 mol干基气体变换后为(1+ y · x )mol的干基气体,则:
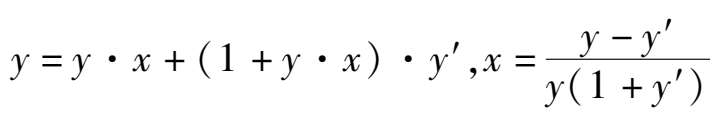
当反应达到平衡时,各组分的平衡组成分别为 y a - y a x , y b - y a x , y c + y a x 和 y d + y a x ,所以变换反应平衡常数可表示为:
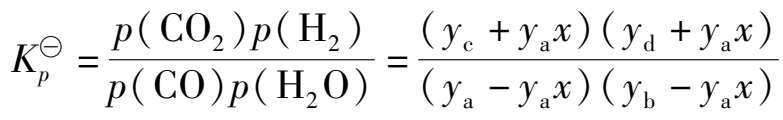
(1.3.1)
上述包含了水蒸气的气体组成,则称为湿基组成。在干基组成和湿基组成之间可通过汽气比,即 1 mol干原料气体中水蒸气的摩尔数,进行换算。如测得原料气的干基含量并已知汽气比,即可求得一氧化碳变换率 x 。温度越低,水碳比越大,平衡变换率越高。
因平衡变换率随温度升高而降低,但反应速度却随温度升高而增加,为了既保持较高的反应速度,又尽量提高平衡变换率,工业上常采用先较高温度(400~450℃)变换后较低温度(200~250℃)变换的两段变换工艺。前者称为中温变换或中变,后者称为低温变换或低变。
2)变换反应动力学
在有关一氧化碳变换反应动力学的研究工作中,由于使用催化剂的性能、型号的差异以及实验条件不同,整理出来的变换动力学方程式各异。在工艺计算中,较常用的动力学方程式有三类:
(1)一级反应
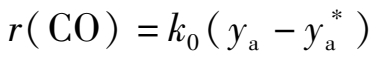
(1.3.2)
式中
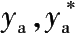 ——一氧化碳的瞬时含量与平衡含量,摩尔分数;
——一氧化碳的瞬时含量与平衡含量,摩尔分数;
k 0 ——反应速率常数,m 3 /(m 3 ·h);
r (CO)——反应速率,以单位体积催化剂单位时间反应的CO在标态下的体积表示,m 3 /(m 3 ·h)。
其等温积分式为:
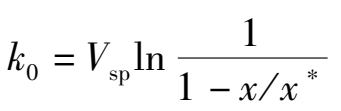
(1.3.3)
或
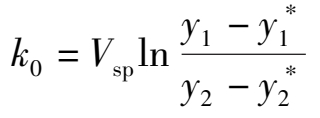
(1.3.4)
式中 V sp ——湿原料气空速,h -1 ;
x , x *——一氧化碳的变换率和平衡变换率;
y 1 , y 2 ——进、出口气体中一氧化碳含量,摩尔分数;
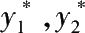 ——进、出口气体中一氧化碳的平衡含量,摩尔分数。
——进、出口气体中一氧化碳的平衡含量,摩尔分数。
(2)二级反应
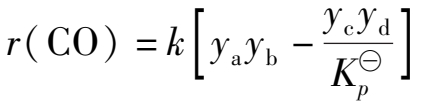
(1.3.5)
式中 y a , y b , y c , y d ——CO,H 2 O,CO 2 和H 2 的瞬时含量,摩尔分数;
k ——与温度有关的速率常数。
对国外G-3A型中变催化剂, k = exp(15.99 -4 900/ T );国产中变催化剂,B104 型(即C 4~2 ), k = exp(17.94 -6 300/ T ); B106 型(C 6 ) k = exp(17.88 -5 920/ T );B109 型(C 9 ) k =exp(16.70 -4 940/ T );对低变催化剂,G-66 型 k = exp(15.92 -3 917/ T );G-66B型 k = exp(12.88 -1 856/ T )。
对于变换反应,一般认为内扩散的影响不能忽略。内表面利用率不仅与催化剂的尺寸、结构以及反应活性有关,而且与操作温度和压力等因素有关。通过计算发现,对同一尺寸的催化剂,在相同压力下,提高操作温度,则一氧化碳扩散速度也相应增加,但在催化剂内表面上反应的速度常数增加更为迅速,总的结果是随温度升高,内表面利用率降低。在相同的温度和压力下,小颗粒的催化剂具有较高的内表面利用率,这主要是因为催化剂的尺寸越小,毛细孔的长度越短,内扩散阻力越小的缘故。对同一尺寸的中变催化剂,在温度相同时提高压力,反应速度会增大,而一氧化碳有效扩散系数又显著减小,故内表面利用率降低。
3)催化剂
(1)中变催化剂
只有在催化剂存在时,一氧化碳变换反应速度才能满足工业生产的要求。目前广泛应用的中变催化剂是以Fe 2 O 3 为主体,以Cr 2 O 3 为主要添加物的多成分铁铬系催化剂。
近年来,随着重油部分氧化法的发展,所制得的原料气含硫量高,又逐渐开始采用耐高硫的钴钼系变换催化剂。国产中变催化剂的性能如表 1.4 所示。
表 1.4 国产铁铬系中变催化剂的性能
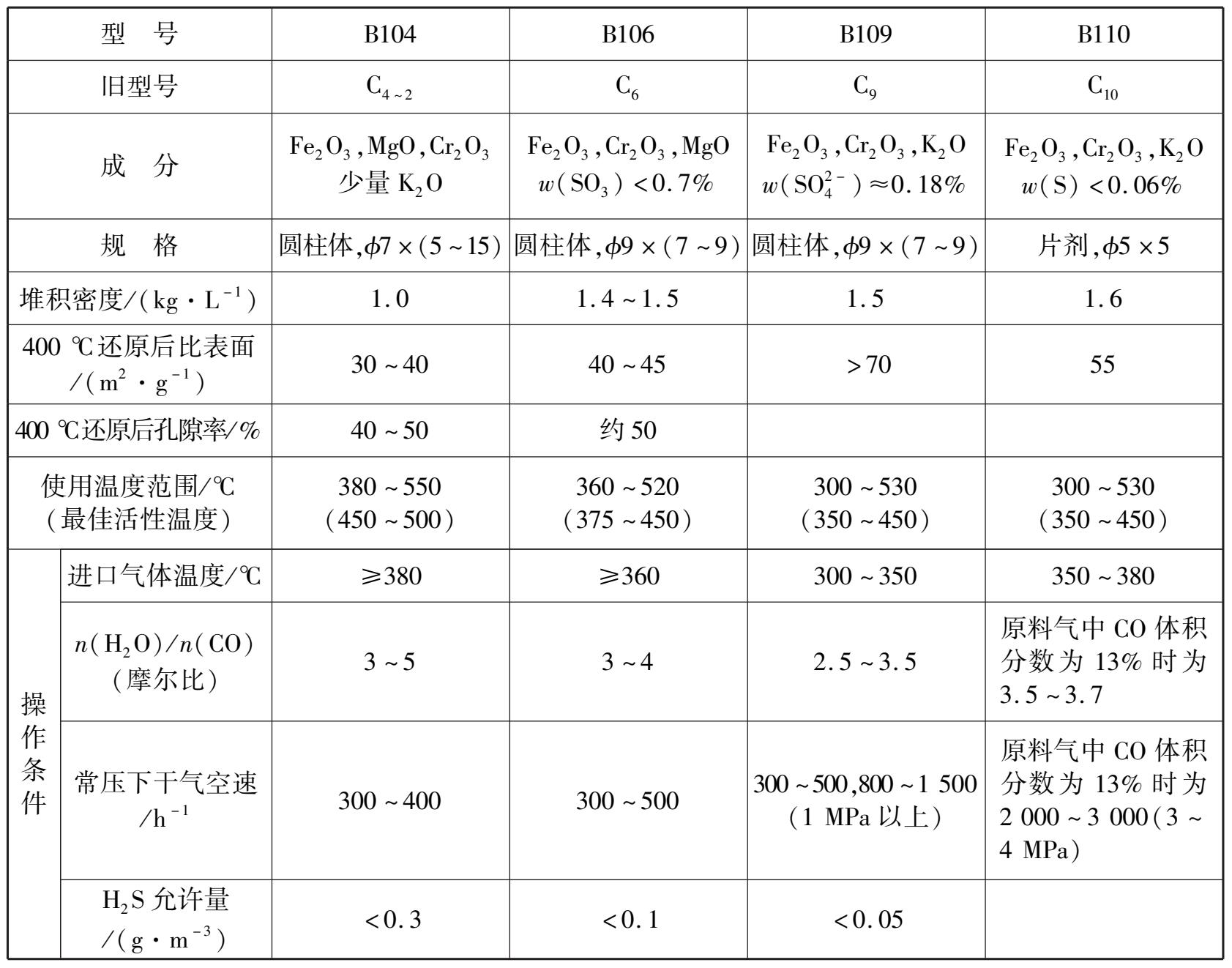
铁铬系催化剂中 w (Fe 2 O 3 )一般为 80%~90%, w (Cr 2 O 3 )为 7%~11%,并含有K 2 O(K 2 CO 3 ),MgO及Al 2 O 3 等成分。在催化剂的各种添加物中,以Cr 2 O 3 最为重要。它的主要作用是将活性组分Fe 2 O 3 分散,使之具有更细的微孔结构和较大的比表面积,防止Fe 3 O 4 的结晶成长,使催化剂耐热性能提高,延长使用寿命,提高催化剂的机械强度,抑制析碳副反应等。添加K 2 CO 3 也能提高催化剂的活性。单独添加极少量的K 2 CO 3 就有一定促进效果,如同时添加Cr 2 O 3 和K 2 CO 3 ,则效果更佳。添加MgO及Al 2 O 3 虽不能提高催化剂的活性,但可增加催化剂的耐热性,而且MgO还具有良好的抗硫化氢的能力。铁铬系催化剂中,Fe 2 O 3 对一氧化碳变换反应无催化作用,需要还原成Fe 3 O 4 才具有活性。在生产中,通常用含氢或CO的气体进行还原,其反应如下:
3Fe 2 O 3 +CO═2Fe 3 O 4 +CO 2
3Fe 2 O 3 +H 2 ═2Fe 3 O 4 +H 2 O
近年来开发的中变钴钼催化剂,以氧化铝为载体,有效组分为氧化钴(3%~3.5%)和三氧化钼(10%~15%),并加入少量碱金属氧化物。在使用前,用含硫化氢的气体硫化,实际起催化剂作用的是硫化钴和硫化钼。钴钼催化剂不受硫化物的毒害。它的活性温度比铁铬催化剂低,在 250~400℃就可使用,机械强度大于铁铬催化剂,不会将硫化氢还原成单质硫,从而不至于粘附堵塞设备。使用方法比铁铬催化剂简单,铁铬催化剂在使用前要经过仔细还原,还原条件明显影响催化剂的活性、机械强度和使用寿命。
(2)低变催化剂
目前工业上应用的低变催化剂以氧化铜为主体,还原后的活性组分是细小的铜结晶,在操作温度下极易烧结,比表面积小,从而催化剂活性下降,寿命缩短。为此在催化剂中加入氧化锌和氧化铝,使微晶铜有效地被分隔开来不致长大,从而提高了催化剂的活性和热稳定性。低变催化剂中 w (CuO)为 15.3%~31.2%, w (ZnO)为 32%~62.2%, w (Al 2 O 3 )为 30%~40.5%。
使用低变催化剂前,需用氢或一氧化碳将其还原成具有活性的微晶铜,其反应如下:
CuO+H 2 ═Cu+H 2 O(g)
CuO+CO ═Cu+CO 2
氧化铜还原是强烈的放热反应,因此必须严格控制还原条件,将催化层温度控制在 230℃以下。硫化物和氯化物是低变催化剂的主要毒物,硫化物使低变催化剂永久性中毒。催化剂中硫的质量分数为 0.1%时,变换率下降 10%;硫的质量分数为 1%时,变换率下降 80%。硫化物除了来自原料气以外,还可能来自中变催化剂,所以在中变催化剂进行还原时,应注意“放硫”完全,以免低变催化剂中毒。在一些装置中,于低变催化剂上部装入ZnO作为防硫保护层。氯化物对低变催化剂的作用较硫化物大 5~10 倍,它能破坏催化剂结构造成永久性中毒。氯化物主要来源于水蒸气或变换炉冷激用的冷凝水。为了保护催化剂,要求蒸汽中氯的体积分数低于 3.0 × 10 -8 。
4)变换过程工艺条件
(1)温度
变换反应是可逆的放热反应,因此存在最佳反应温度。在原始气体组成和催化剂一定的条件下,变换反应正逆两向的反应速度均随温度的升高而增加,而平衡常数则随温度的增高而减少。在低温阶段反应远离平衡时,增加温度使变换过程总速度增加;当温度增至某一值时,反应速度达到最大;超过这一温度后,由于平衡的限制和逆反应的影响,增加温度总的反应速度反而下降。对应的最大反应速度时的温度称为在该条件下的最佳反应温度。随着反应的进行,气体组成随时都在变化,每一瞬时组成都对应着一个最佳温度。过程之初,一氧化碳变换率低,最佳温度高;随着反应的进行,变换率逐渐增加,最佳温度逐渐下降,形成一定的最佳温度分布。把不同变换率下的各最佳点连接成曲线,称为最佳温度分布曲线。如果变换按最佳温度分布曲线进行,则整个过程的反应速度最大,在相同的生产力下所需催化剂用量最少。最佳温度 T o 与平衡温度 T e 的关系为:
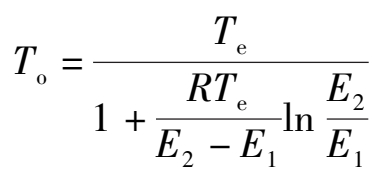
(1.3.6)
式中 E 1 , E 2 ——正逆反应的活化能,J/mol。
不同变换率 x 时的最佳温度 T o 如图 1.17 所示。
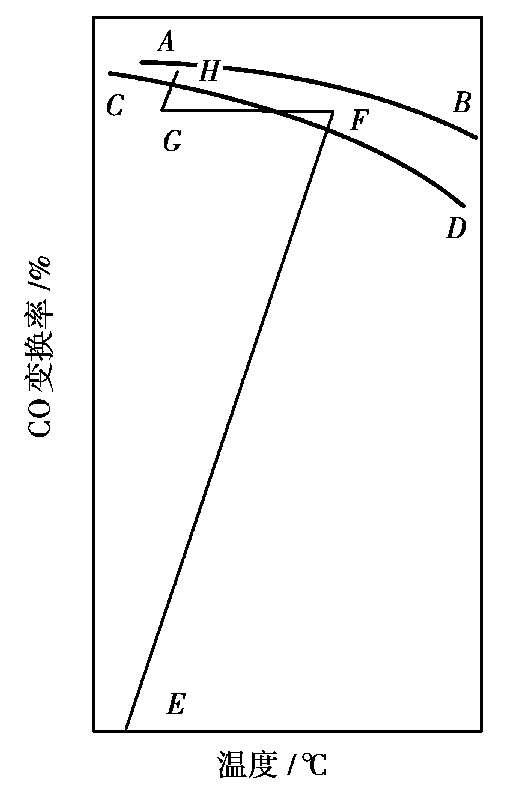
图 1.17 CO变换过程的
T-x
图
AB
—平衡温度线;
CD
—适宜温度线;
E
—进入第一段催化剂层的状态;
F
—离开第一段催化剂层的状态;
G
—进入第二段催化剂层的状态;
H
—离开第二段催化剂层的状态;
FG
—中间冷却过程
图 1.17 是一个两段绝热反应、段间间接换热的 T-x 操作状况图。 EF , GH 分别为一、二段绝热反应操作线, FG 为段间间接换热降温线,绝热操作线方程可由热量衡算导出。如采用平均温度时的反应热(-Δ H R )和平均热容 C P 计算,可得出:
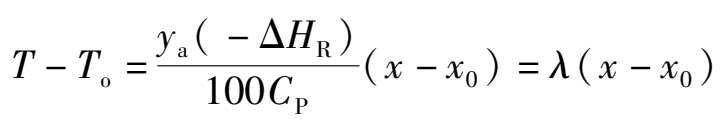
(1.3.7)
式中 λ ——绝热温升。
显然,绝热反应分段越多,则操作越接近最佳反应温度曲线,但流程越复杂。根据原料气中的CO的体积分数,一般多将催化剂床层分为一段、二段或多段,段间进行冷却。冷却的方式有两种:一是间接换热式,用原料气或饱和蒸汽进行间接换热;二是直接冷激式,用原料气、水蒸气或冷凝水直接加入反应系统进行降温。
(2)压力
压力对变换反应的平衡几乎没有影响,但提高压力将使析碳等副反应易于发生,所以单就平衡而言,加压并无好处。但从动力学角度分析,加压可提高反应速度,因为变换催化剂在加压下比常压下活性更高(图 1.18)。加压变换设备体积小,布置紧凑。加压变换过程的湿变换气中水蒸气冷凝温度高,有利于热能回收。一般小型氨厂操作压力为0.7~1.2 MPa,中型氨厂为 1.2~1.8 MPa,以煤为原料,纯氧气化的大型氨厂,压力可达 5.2 MPa,以烃类为原料的大型合成氨厂压力为 3.0 MPa。
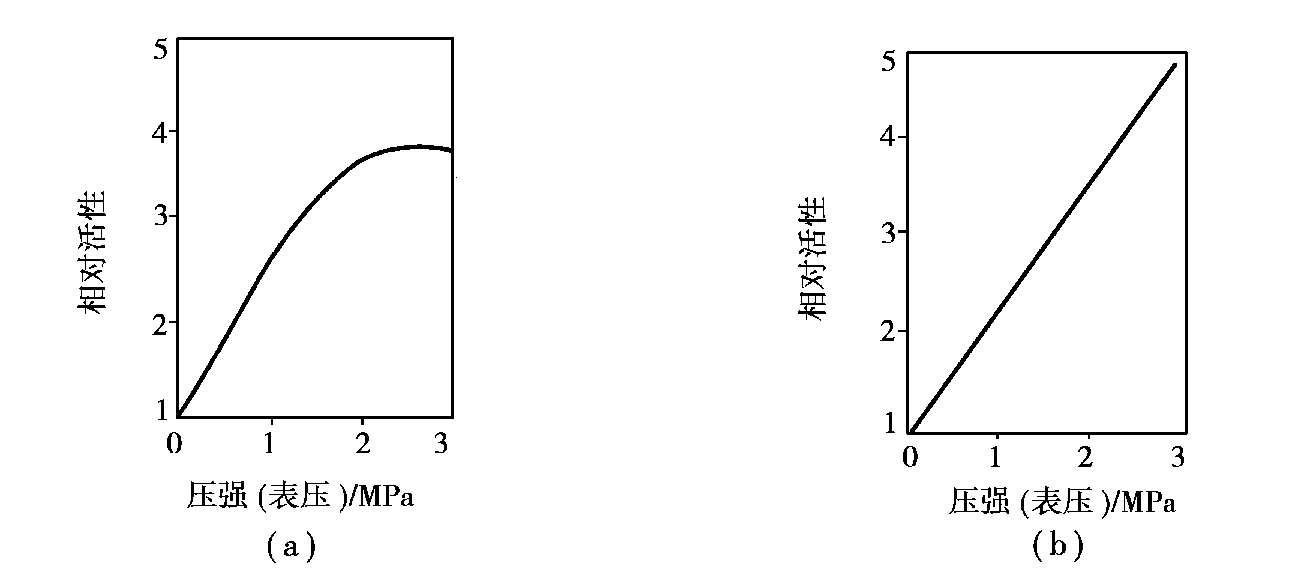
图 1.18 加压下催化剂的活性
(a)某中变催化剂;(b)某低变催化剂
(3)水蒸气比例
水蒸气比例是指水蒸气与原料气中一氧化碳的摩尔比或蒸汽与干原料气的摩尔比。增加水蒸气用量,一氧化碳平衡变换率提高,反应速度加快,可阻止催化剂还原,避免析碳及生成甲烷的副反应。同时可使原料气中一氧化碳体积分数下降,绝热温升 λ 减小,所以改变水蒸气用量是调节床层温度的重要手段。但是水蒸气用量也不宜过高,否则不仅蒸汽消耗量增加,而且床层压降太大,反应温度难以维持。中变水蒸气比例一般为 3~5。
5)变换反应的工艺流程
综上所述,一氧化碳变换工艺的流程安排应做如下考虑。若一氧化碳体积分数较高,则应采用中温变换,因为中变催化剂操作温度范围较宽,而且价廉、寿命长,大多数合成氨原料气中CO均高于 10%,故都可先通过中变除去大部分一氧化碳。对一氧化碳体积分数高于 15%者,一般可考虑适当分段,段间进行冷却降温,尽量靠近最适宜温度操作。其次,根据原料气的温度与湿含量情况,考虑适当预热和增湿,合理利用余热。如允许变换气中残余CO的体积分数在 3%左右,则只采用中变即可。如要求在 0.3%左右,则将中变和低变串联使用。下面介绍两种典型的变换工艺流程。
(1)中变-低变串联流程
以烃类蒸汽转化制氢为例(图 1.19),CO的体积分数为 13%~15%的原料经废热锅炉(1)降温,在压力 3 MPa、温度 370℃下进入高变炉(2),因原料气中湿含量较高,一般不需添加水蒸气,反应后气体中一氧化碳降至 3%左右,温度 425~440℃,通过高变废热锅炉(3)冷却到330℃。高变气继续进入甲烷化炉进气预热器(4),温度降至 220℃后进入低变炉(5),出炉温度 240℃左右,残余一氧化碳的体积分数降至 0.3%~0.5%。低变气还可进一步回收余热,气体离开变换系统后进入二氧化碳吸收塔。
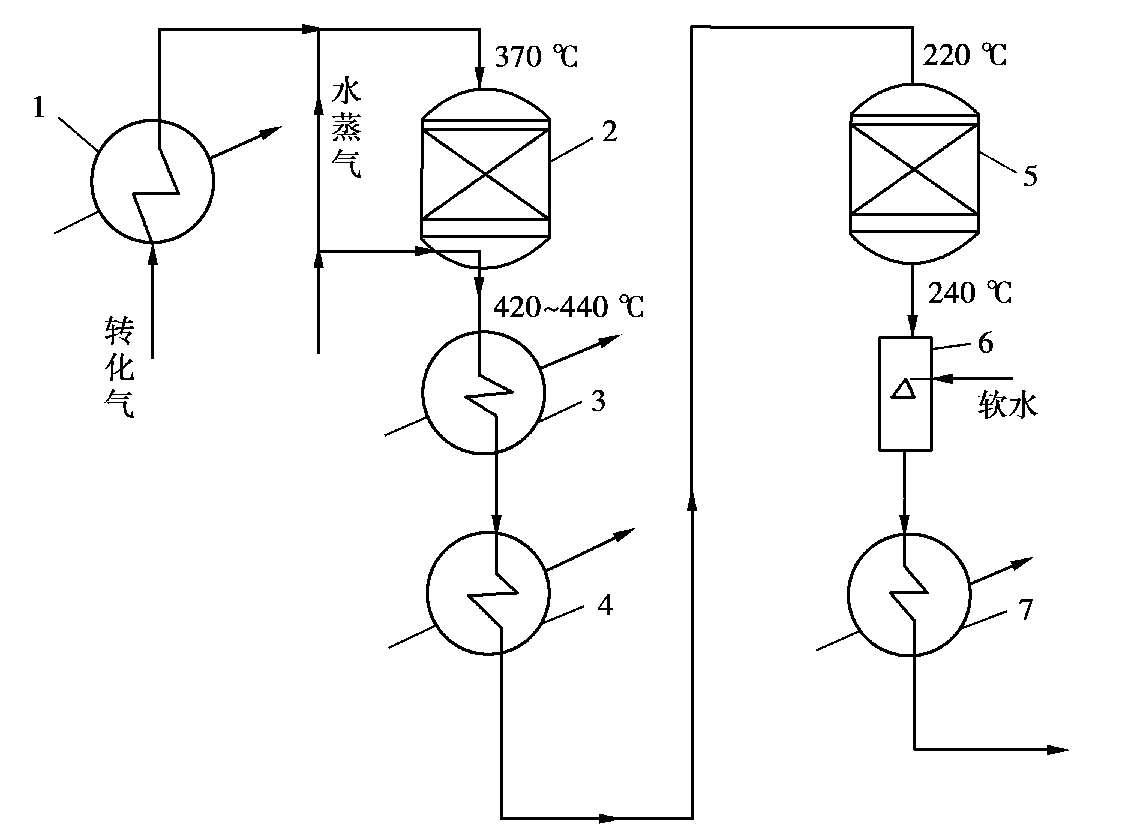
图 1.19 一氧化碳中变-低变串联流程
1—废热锅炉;2—高变炉;3—高变废热锅炉;4—甲烷化炉进气预热器;5—低变炉;6—饱和器;7—釜液再沸器
(2)多段变换流程
多段变换流程如图 1.20 所示。原料气进入饱和塔下部与水循环泵打来并经水加热器加热的热水逆流接触,气体被加热到 160~190℃,水被冷却至 135~150℃。然后气体由塔顶逸出,在管道内与外供的高压蒸汽混合后,经换热器和中间换热器加热至 400℃左右进入变换炉一段。这里,约有 80%的一氧化碳被变换成氢,反应热使气体温度升至 520℃左右,引出到中间换热器降温至 420℃后,进入变换炉二段。此时,气体中一氧化碳体积分数降至 3.5%以下,温度约 430℃,由炉底部逸出,依次经换热器、水加热器、热水塔降温至 160℃后,送下一工段处理。
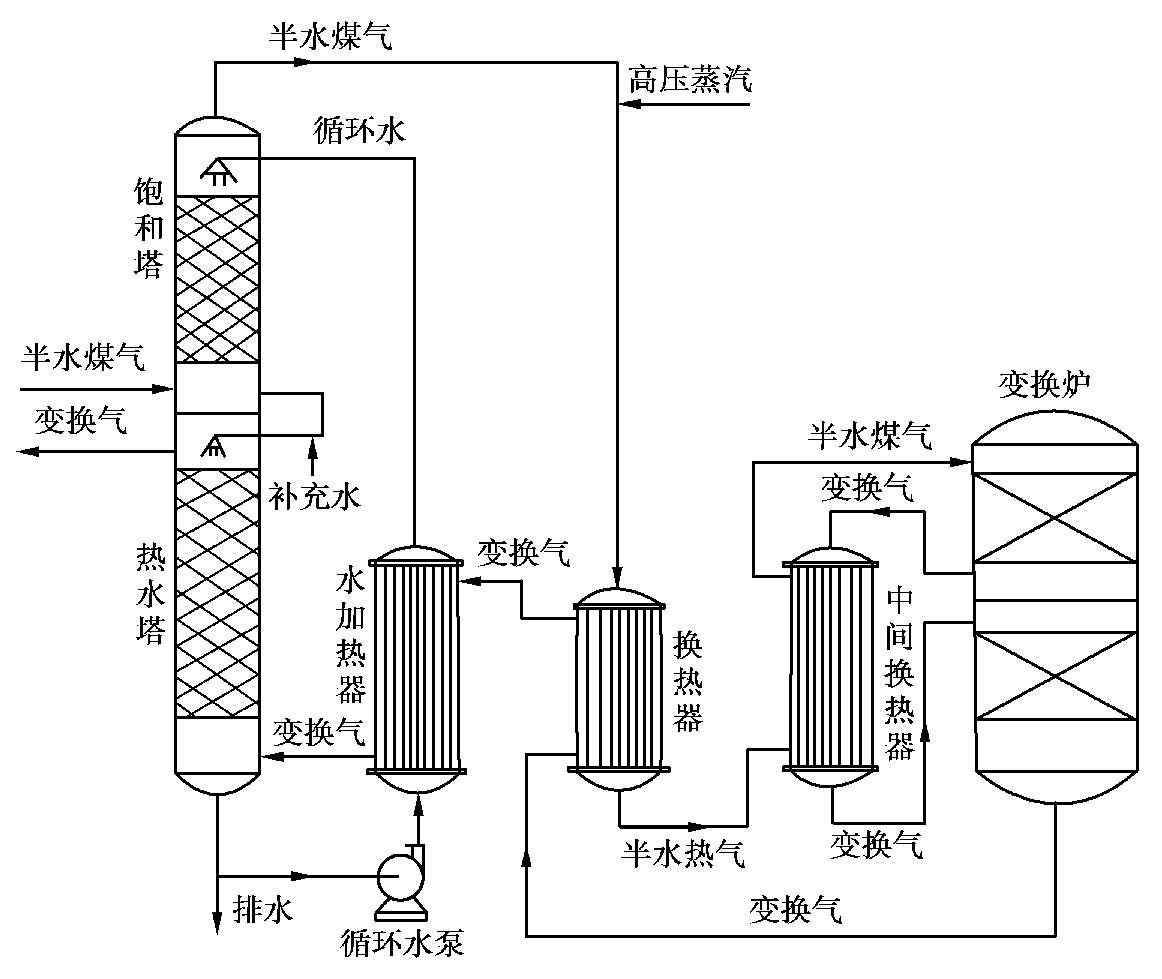
图 1.20 多段变换流程
1.3.3 二氧化碳的脱除
经变换后的气体中含有较多的二氧化碳,回收二氧化碳可作为制造尿素、纯碱、碳酸氢铵的重要原料,习惯上把脱除和回收二氧化碳的过程称为“脱碳”。
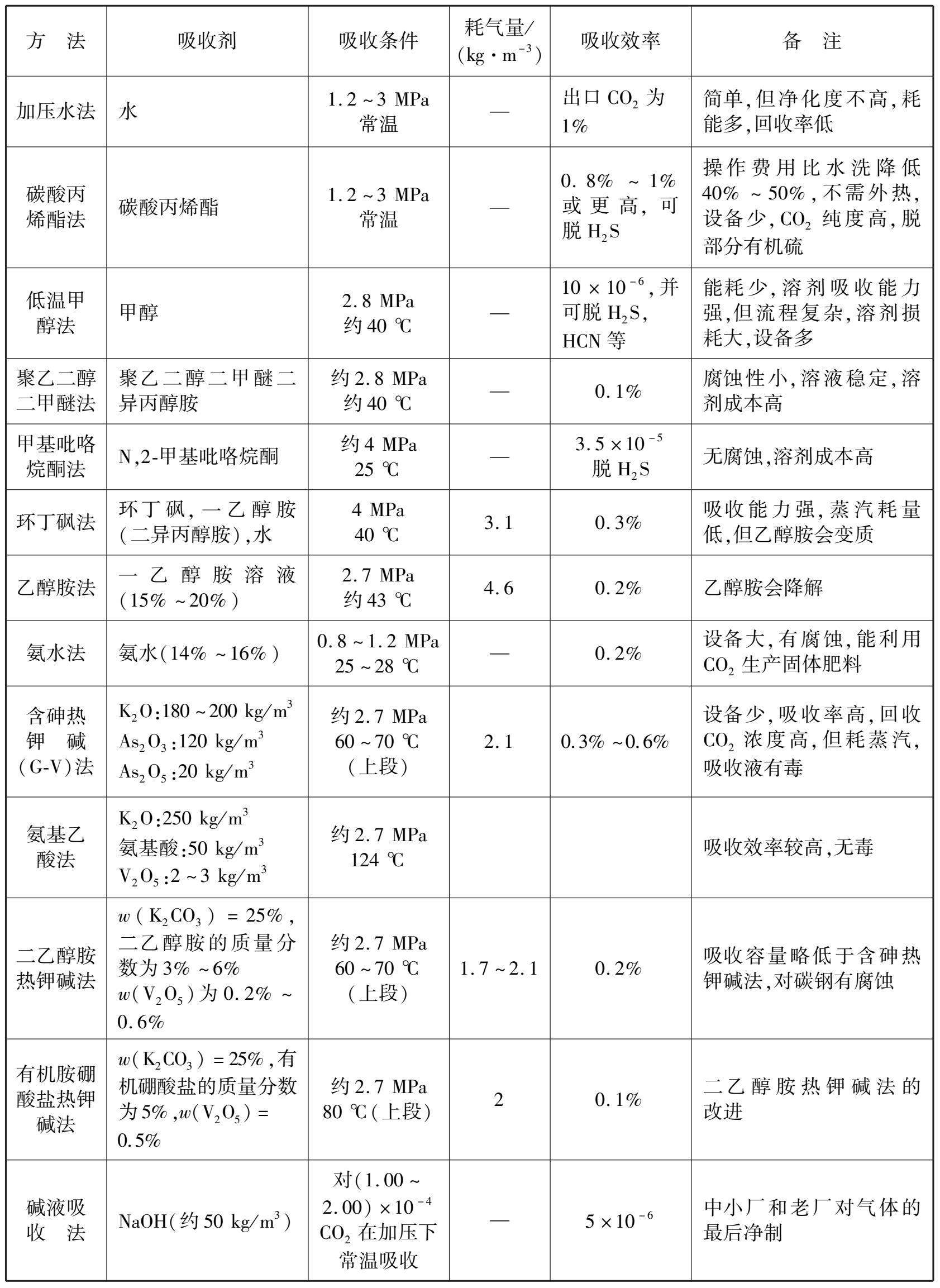
像脱硫一样,二氧化碳的脱除有多种方法,应根据原料气的组成、净制要求、前后工序关系、生产规模,环境安全等因素综合考虑,这些方法的概况列于表 1.5。
表 1.5 变换气脱二氧化碳的方法
1)苯菲尔法脱碳
二乙醇胺催化热钾碱法脱除CO 2 称为苯菲尔法脱碳。
碳酸钾水溶液呈强碱性,吸收CO 2 的化学反应式为:
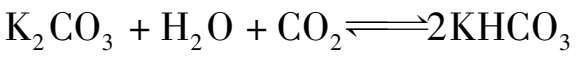
为了提高其反应速度以及增加KHCO 3 的溶解度,吸收在较高温度(105~130℃)下进行,故称热碳酸钾法。同时在此温度范围内,吸收温度与再生温度基本相同,可节省吸收液再生时所消耗的热量,但在此温度下吸收CO 2 的速度仍然太慢,且吸收液对设备腐蚀也很严重。
脱碳后气体的净化度与K 2 CO 3 水溶液中CO 2 的平衡分压有关。溶液CO 2 的平衡分压越高,达到平衡后气体中残余的CO 2 越高,气体的净化度就越低。此外,K 2 CO 3 水溶液中CO 2 的平衡分压还与K 2 CO 3 溶液的浓度、溶液中K 2 CO 3 转变成KHCO 3 的转化率以及吸收时的温度有关,温度升高,则CO 2 的平衡分压增大。图 1.21 为CO 2 在苯菲尔脱碳溶液上的平衡分压。
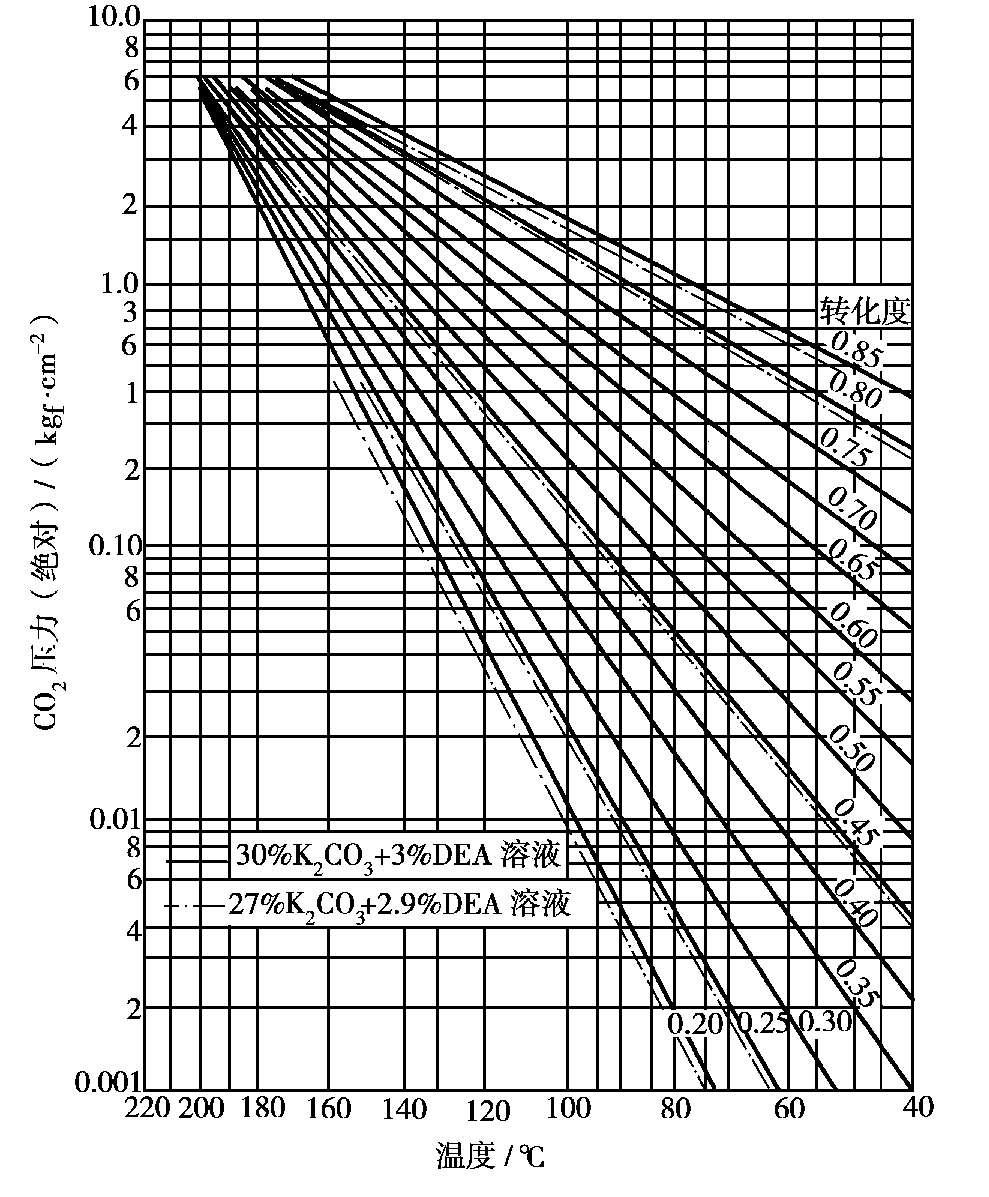
图 1.21 苯菲尔脱碳溶液上CO 2 的平衡分压
在K 2 CO 3 的溶液中加入少量活化剂,如二乙醇胺[CH 2 CH(OH) 2 NH 2 ,简称DEA]可以改变吸收反应历程,从而大大加快吸收CO 2 的速度,同时还可降低液面上CO 2 的平衡分压,使脱碳气的净化度提高。表 1.6 列出了DEA对碳酸钾溶液平衡分压和吸收系数的影响。
表 1.6 DEA对碳酸钾溶液平衡分压和吸收系数的影响
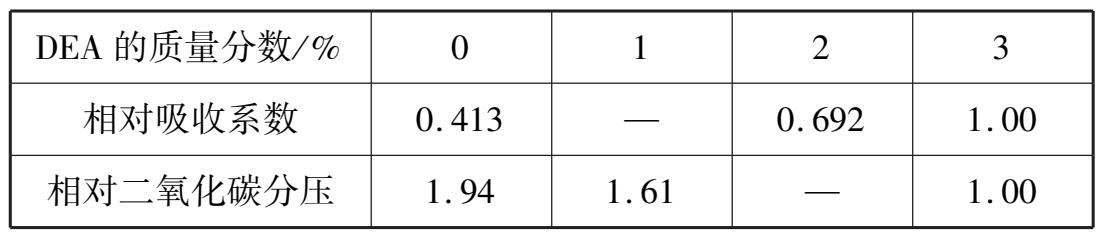
这种加入了DEA的碳酸钾溶液在吸收脱除CO 2 的同时,还能除去原料气中的H 2 S等酸性组分。
碳酸钾溶液吸收CO 2 后,K 2 CO 3 转变为KHCO 3 ,溶液的pH值减小,吸收能力下降。需要将溶液再生,释放出CO 2 ,使溶液恢复吸收能力。再生反应为:
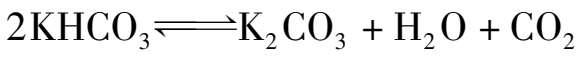
加热有利于碳酸氢钾的分解。为了使CO 2 从溶液中充分解吸出来,一般将溶液加热至沸点,使大量的水蒸气逸出,有利于降低气相中CO 2 的分压,起到气提作用。再生时压力越低,对再生的效果越好。但为了简化流程和便于将解吸出的CO 2 送到后序工段,其压力一般高于大气压约 0.015 MPa,而再生温度为该压力下溶液的沸点温度。再生温度和再生压力与溶液的组成有关。
在实际操作过程中,考虑到在高温下高浓度碳酸钾溶液对设备的腐蚀以及在低温下易析出结晶的特点,通常在吸收时,采用碳酸钾的质量分数为 25%~30%,活化剂DEA的质量分数为 2.5%~5%。溶液中一般还需加入偏钒酸钾(KVO 3 )或V 2 O 5 为缓蚀剂,让其与铁作用,在设备表面形成一层氧化铁保护膜,使设备免受热钾碱溶液和CO 2 的腐蚀。溶液中KVO 3 的质量分数为 0.6%~0.9%,并要求溶液中五价钒的质量分数为总钒的 20%以上。此外溶液中还需加入微量的有机硅酮类、聚醚类以及高级醇类等有机化合物作为抑制发泡的消泡剂。
苯菲尔法脱除CO 2 的工艺流程如图 1.22 所示。低变气用水淬冷到露点,进入低变气再沸器,放出大量冷凝热作为再生热源。喷水淬冷的目的是降低气体进再沸器的温度,减少壳侧热碱液的腐蚀,同时又能提高再沸器的传热效果。低变气体进分离罐分出冷凝水,然后入吸收塔底部,从下而上与热碱液逆流接触,CO 2 被吸收后,气体中还残余约 1.0× 10 -3 的CO 2 。从塔顶出来的气体进入液滴分离罐,除去夹带溶液,溶液回到贮槽,由液滴分离罐出口的气体到下一工段甲烷化。
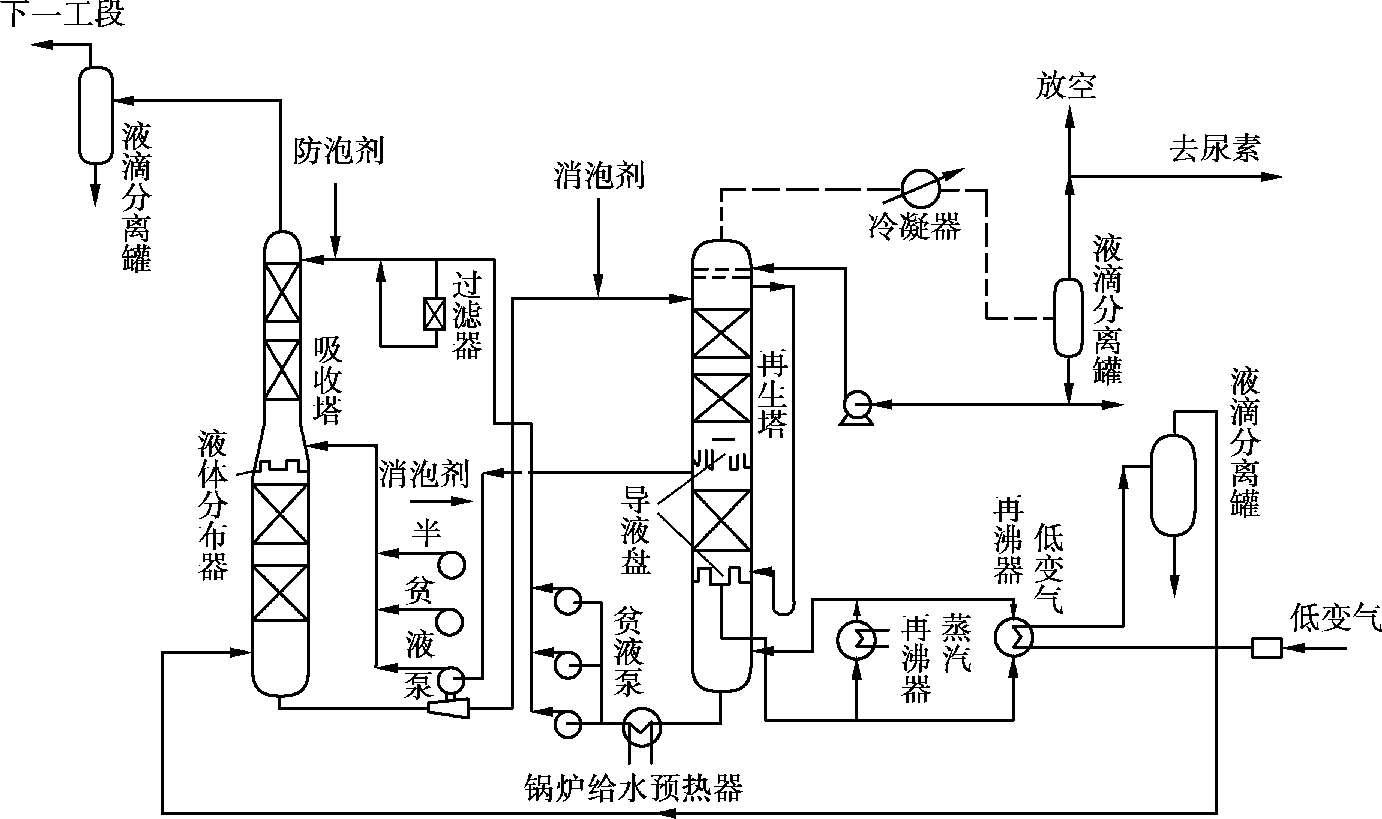
图 1.22 苯菲尔法的工艺流程图
目前大型氨厂苯菲尔脱碳工序普遍采用两段吸收、两段再生流程。吸收塔有两个进液口,从塔中部进入的是来自再生塔中部的半贫液,贫液由再生塔底部流出,经锅炉给水预热器,进入吸收塔顶部,自上而下再与半贫液汇合。一部分贫液进吸收塔前,先入过滤器除去杂质,以防止起泡。入过滤器的液量约占贫液总量的1/10。吸收塔底部的富液利用本身的压力到再生塔顶部,由于吸收塔压力很高,所以富液可以驱动一台水力透平回收能量。富液经水力透平减压,在再生塔顶就闪蒸出一些CO 2 。吸收液自上而下与热气体(水蒸气与CO 2 的混合物)逆流接触,溶解的CO 2 不断放出。半贫液从再生塔中抽出,入吸收塔中部。小部分溶液(约占总量1/4)再进一步再生,最后流入变换气再沸器和蒸汽再沸器,保持沸腾状态,使溶液中CO 2 脱到规定范围,返回再生塔底部。贫液经锅炉给水预热器被锅炉给水冷却,送吸收塔顶。由再生塔出来的CO 2 经洗涤后,入冷凝器冷却,并进液滴分离罐分出冷凝水后,送往氨加工车间。在吸收塔顶、中部和再生塔顶部,设有消泡剂注入口,根据需要加入。
2)含砷热钾碱法(G-V法)脱碳
含砷热钾碱法是往碳酸钾溶液中添加三氧化二砷作为助剂,并起催化作用。碳酸钾与三氧化二砷反应生成砷酸二氢钾:
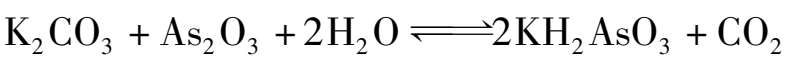
含砷钾碱溶液吸收CO 2 时的反应为:
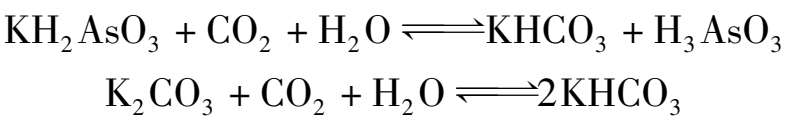
含砷热钾碱溶液吸收CO 2 的平衡如图1.23 所示,该体系的CO 2 平衡分压比相应的普通碳酸钾溶液体系要低得多。
图 1.23 As 2 O 3 的质量浓度为 120 kg/m 3 的砷钾碱液吸收CO 2 的平衡
钾碱液中添加三氧化二砷后,不仅提高了钾碱液对CO 2 的吸收量,也加快了吸收CO 2 的速率,使净制气中CO 2 下降。以CO 2 的体积分数为 25%的变换气在12 MPa和 75℃下吸收为例,净制气中CO 2 质量分数与碱液(碱液折算为K 2 O的质量浓度为200kg/m 3 )中添加三氧化砷的量间的关系如图 1.24所示,图中转折点三氧化二砷质量浓度在 30kg/m 3 左右。添加三氧化二砷也提高了吸收富液再生的速率,可以节省蒸汽,再生时蒸汽消耗量与碱液中三氧化二砷质量浓度的关系如图 1.25 所示。
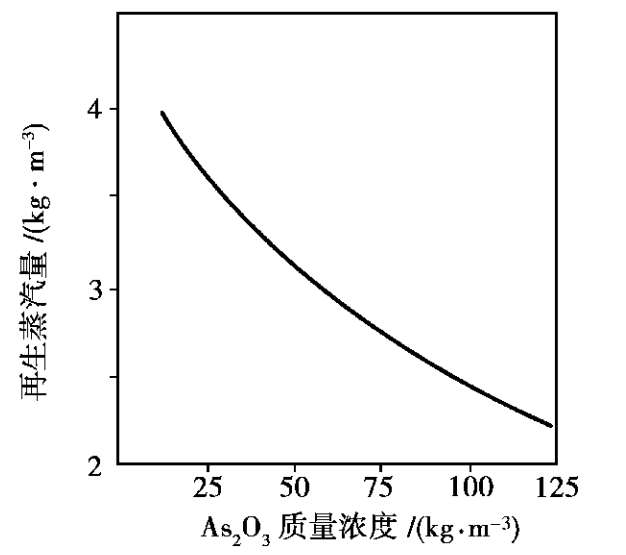
图 1.25 As
2
O
3
质量浓度对再生用蒸汽量的影响[进气中
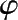 (CO
2
)= 28%,出气中
(CO
2
)< 1%]
(CO
2
)= 28%,出气中
(CO
2
)< 1%]
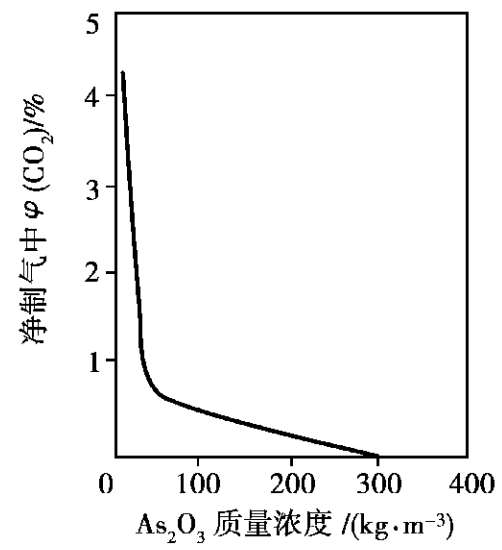
图 1.24 As
2
O
3
质量浓度对净制程度的影响[进气中
(CO
2
)= 25%,1.2 MPa]
综合以上关系,常用的砷钾碱液的组成为:K 2 O的质量浓度为 200 kg/m 3 ,As 2 O 3 的质量浓度为 120 kg/m 3 ,As 2 O 5 的质量浓度为 20 kg/m 3 ,1 m 3 碱液吸收CO 2 的能力为 20~25 m 3 。
含砷热碱法提高了吸收和再生速率,使吸收塔和再生塔比普通热钾碱法小得多,并在80~90℃或更低的条件下进行,净化气中CO 2 质量分数小于 0.2%,含砷热钾碱溶液对碳钢腐蚀小,大多数设备可以用普通碳钢制作。
含砷热钾碱法的工艺流程如图 1.26 所示。变换气经过变换气煮沸器由吸收塔下部通入,与碱液冷却器来的温度为 100℃左右的碱液逆流相遇,气体中CO 2 即被碱液吸收。碱洗后的气体由塔顶出来,经气体冷却器分出其中夹带的水分并适当冷却后,即为碱洗气,送去脱少量CO工段。
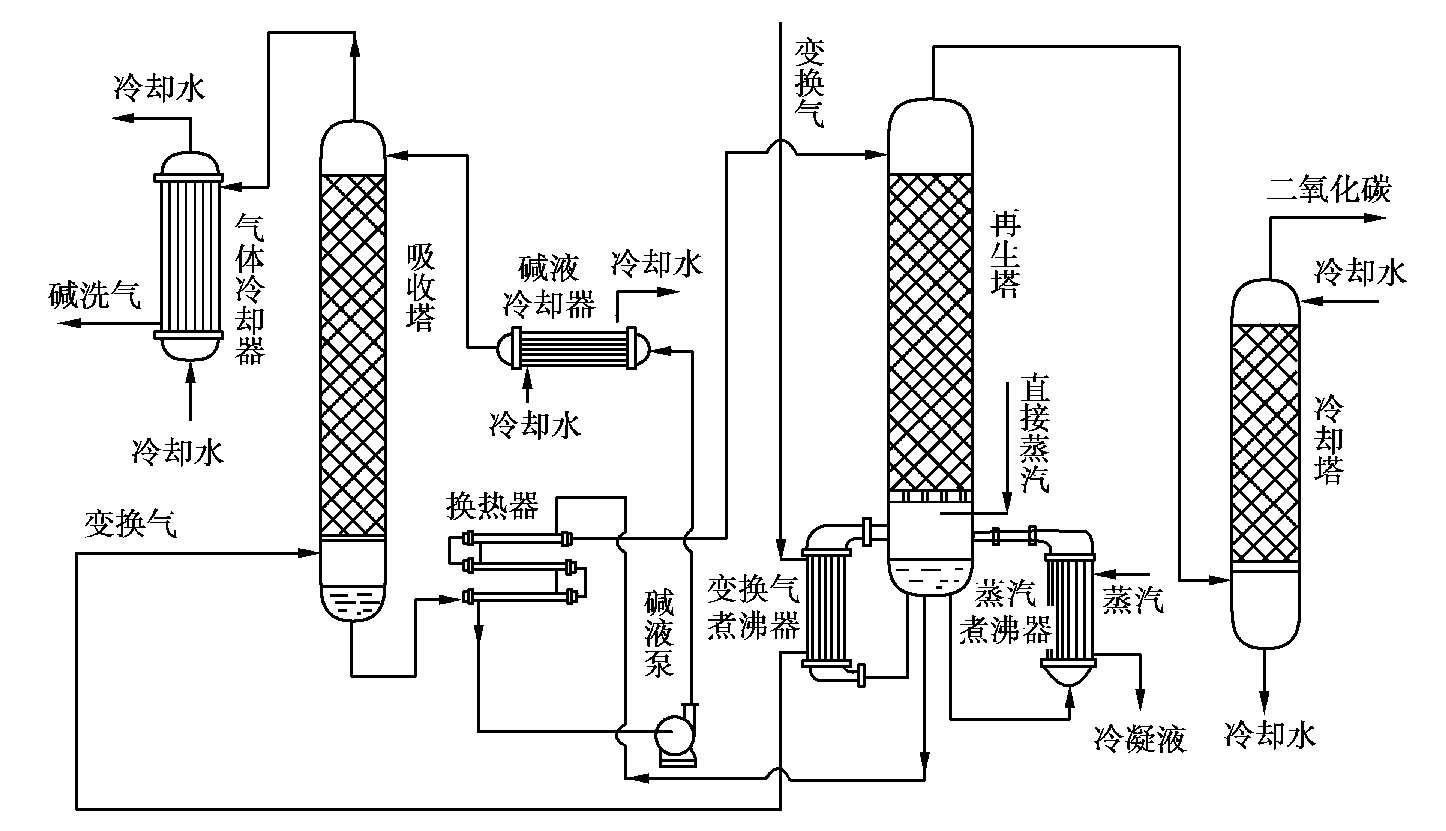
图 1.26 含砷热钾碱法的工艺流程
从吸收塔下部出来的碱液经换热器后进入再生塔上部,具有 105~110℃的待再生碱液,在再生塔中即释放出其中的CO 2 。
为了保证溶液再生完全和回收变换气的热量,采用变换气煮沸器和蒸汽煮沸器来加热塔底的碱液,以供给溶液再生时所需的热量,并有一部分蒸汽直接通入塔中,以补偿再生时水蒸气的消耗。
再生后的碱液用碱液泵升压,经碱液冷却器后,冷却到 100℃左右进入吸收塔循环使用。从再生塔顶出来的CO 2 经冷却塔冷却到常温后,其体积分数在 98%以上,可直接送往尿素车间做原料气用。
1.3.4 少量一氧化碳的脱除
经过变换和脱除二氧化碳,原料气中还含有少量的一氧化碳和二氧化碳。一氧化碳的存在会使合成氨催化剂暂时中毒,因此需在进入合成塔之前脱除,使一氧化碳的质量分数降到规定的 1 ×10 -5 以下。脱除合成氨原料气中残余一氧化碳的方法有铜氨液洗涤法、甲烷化法和液氮洗涤法等。
铜氨液洗涤法是用亚铜盐的溶液在低温和加压下洗涤原料气以吸收一氧化碳,吸收液在减压升温下再生,再生的铜氨液循环使用,再生时释放出的一氧化碳作为再生气回收。通常把铜氨液吸收一氧化碳的操作称为“铜洗”,净化后的气体称为“铜洗气”或“精炼气”。铜氨液洗涤适用于中温变换后一氧化碳含量相对较高的转化气。我国以无烟煤或焦炭为原料的合成氨厂多采用铜洗流程。
甲烷化法是 20 世纪 60 年代开发的新方法,它是在一定的温度和催化剂存在时,使原料气中的一氧化碳与氢作用而生成甲烷。这种方法流程简单,不消耗化学品,催化剂寿命达 2~5年。但甲烷化法消耗原料气中的氢。生成的甲烷虽然对合成氨催化剂无害,可它在合成反应中是惰性气体,降低了反应气中有效组分的浓度。因此此法只适用于低温变换后残余一氧化碳少的转化气。
液氮洗涤法是用液氮在 1.8~2.5 MPa和-190℃左右的条件下洗涤转化气。液氮洗涤时,一氧化碳和甲烷等冷凝入液氮中。吸收后的液氮经过分馏,一氧化碳馏分予以回收,液氮循环使用。液氮洗涤可以同时脱除一氧化碳和甲烷,气体净制程度较高,但需空气分离装置。此法主要用在焦炉气分离以及重油部分氧化、煤富氧气化的制氨流程中。通常把液氮洗涤一氧化碳称为氮洗。下面介绍甲烷化脱除一氧化碳的工艺。
甲烷化法脱一氧化碳是使一氧化碳加氢生成甲烷,其反应如下:
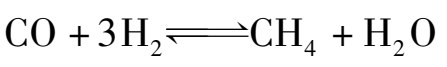
在脱除一氧化碳的同时,CO 2 也进行加氢,其反应如下:
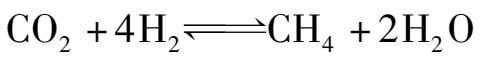
在某种条件下,还会有以下副反应发生:
2CO═C+CO 2
Ni+4CO ═Ni(CO) 4
反应CO与CO 2 甲烷化的平衡常数如表 1.7 所示。由于甲烷化反应是强烈的放热反应,低温有利于正反应,平衡常数较高。在 200~400℃范围内,CO和CO 2 都趋向于生成甲烷,而在大于 600℃的范围,则逆反应易于进行。
表 1.7 甲烷化反应的平衡常数
工业反应温度为 300~400℃时,因 K p 值极大和氢分压高,一氧化碳可认为是全部转化。
需要注意的是,甲烷化反应是强烈的放热反应,在绝热条件下造成反应物系温度的显著升高。在氢氮气中,每 1%CO 2 的绝热温升为 60℃,每 1%CO的绝热温升为 72℃。原料气中CO和CO 2 含量的波动会引起炉温大幅度的变化,应适当控制以免催化剂超温。
在实际生产中,甲烷化反应中一氧化碳分解和羰基镍生成反应是不会出现的。因为甲烷化工业反应温度在 300~400℃,在这个范围内,一氧化碳的分解速度很慢。而羰基镍的反应,温度越低、压力越高对其生成越有利。通过计算发现,在 300~400℃,羰基镍的平衡含量极少。
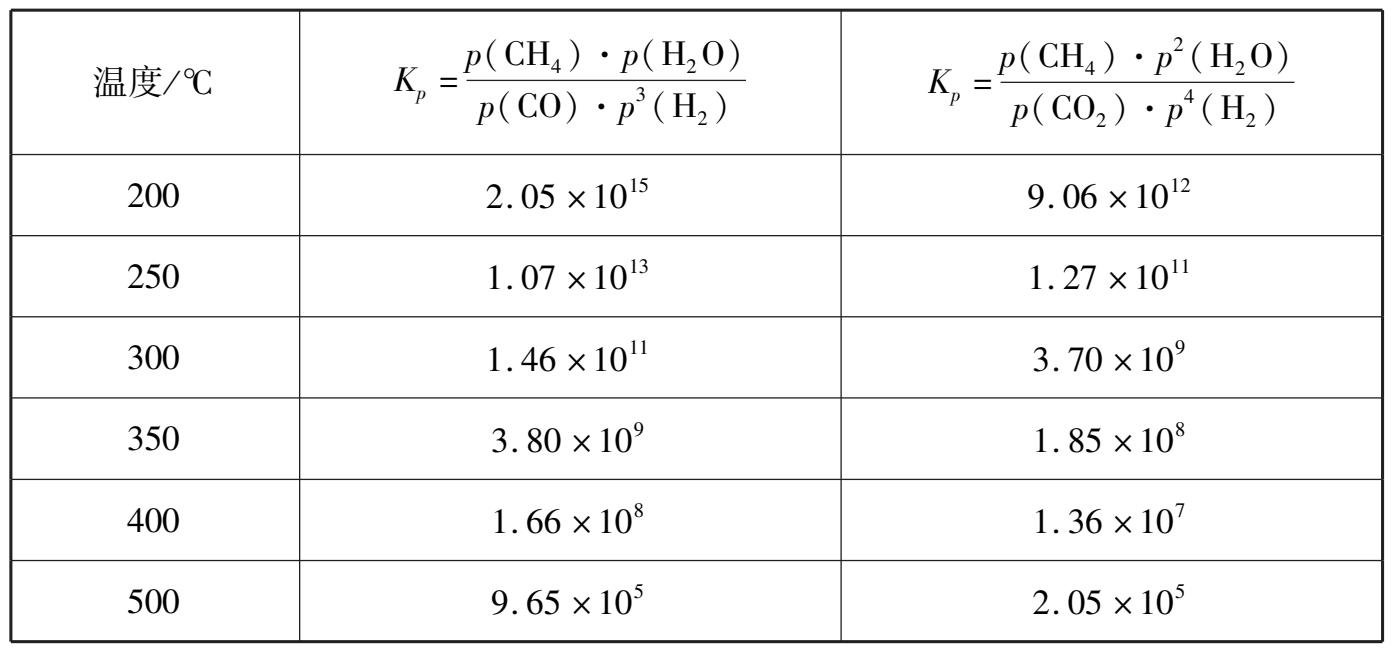
甲烷化反应的动力学研究得出,包括扩散在内的宏观反应速率常数 k 随温度上升而增大,但并不剧烈,以 280℃的 k = 1.0 计,360℃时为 2.50,420℃时为 4.34,可以推测扩散对过程起重要作用。此外,在催化剂上反应速率随操作压力的增大而提高,其关系如图 1.27 所示,但增长值并不大。
图 1.27 操作压力对甲烷化催化剂活性的影响
我国目前使用的甲烷化催化剂是镍催化剂。以镍为主要活性成分,以氧化铝为载体,镍以NiO形式存在。使用前先以氢气或脱碳后的原料气还原,其反应式如下:
NiO+H 2 ═Ni+H 2 O
NiO+CO ═Ni+CO 2
虽然这些还原反应热效应不大,但催化剂一经还原就有活性。用原料气还原时,为了避免床层温升过高,必须尽可能控制碳氧化物体积分数在 1%以下。还原后的镍催化剂会自燃,要防止与氧化性气体接触。当前面工序出现事故,有高浓度的碳氧化合物进入甲烷化反应器时,床层温度会迅速上升,这时应立即切断原料气。
还原后的催化剂不能用含有一氧化碳的气体升温,以防止低温时生成羰基镍。
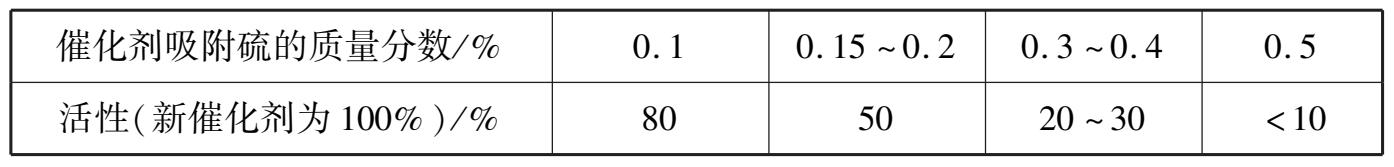
除羰基为甲烷化催化剂的毒物以外,硫、砷和卤素也能使它中毒,即使这些元素微量也会大大降低催化剂的活性和寿命。硫对甲烷化催化剂的危害比对甲烷水蒸气转化催化剂大得多,因其操作温度较低,用无硫气体继续操作也不会使催化剂活性恢复,而对转化催化剂就不然。因此,硫对甲烷化催化剂的危害是累积的。气体含 0.1× 10 -6 的硫可使催化剂的寿命从 5年缩短到 1 年。催化剂吸收 0.5%的硫,活性几乎全部丧失。硫吸附量与活性的关系如表 1.8所示。
表 1.8 甲烷化催化剂中硫吸附量与活性的关系
对砷来讲,吸附量达到 0.1%时,催化剂活性可丧失。在用低温变换的净化流程中,一般不会有硫进入甲烷化反应器,但脱碳系统采用砜胺法、砷碱法时,必须小心操作,以免把这些含有硫和砷的溶液带入。在采用这些方法脱除CO 2 时,为了防止中毒,也可在催化剂床层前加氧化锌作保护剂。
由于前面工序对原料气严格的精制和进口气体中碳氧化物量的限制,在正常情况下不会发生甲烷化催化剂的中毒和烧结,而且本身强度较高,催化剂的寿命可达 3~5 年。取出或者准备再用时,催化剂应该有控制地用预先氧化的方法钝化,其反应式如下:
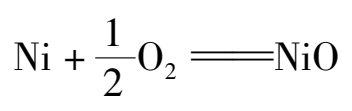
在甲烷化的反应过程中,因考虑到催化剂的升温活化和气体中CO,CO 2 的含量变化,还需采用其他热源,所以甲烷化有两种流程。一种是基本上用甲烷化后的气体来预热甲烷化前的气体,不足量由高变气给出少量热量补充;另一种是完全利用外来热源(高变气或其他)。
现介绍后一种流程,如图 1.28 所示。
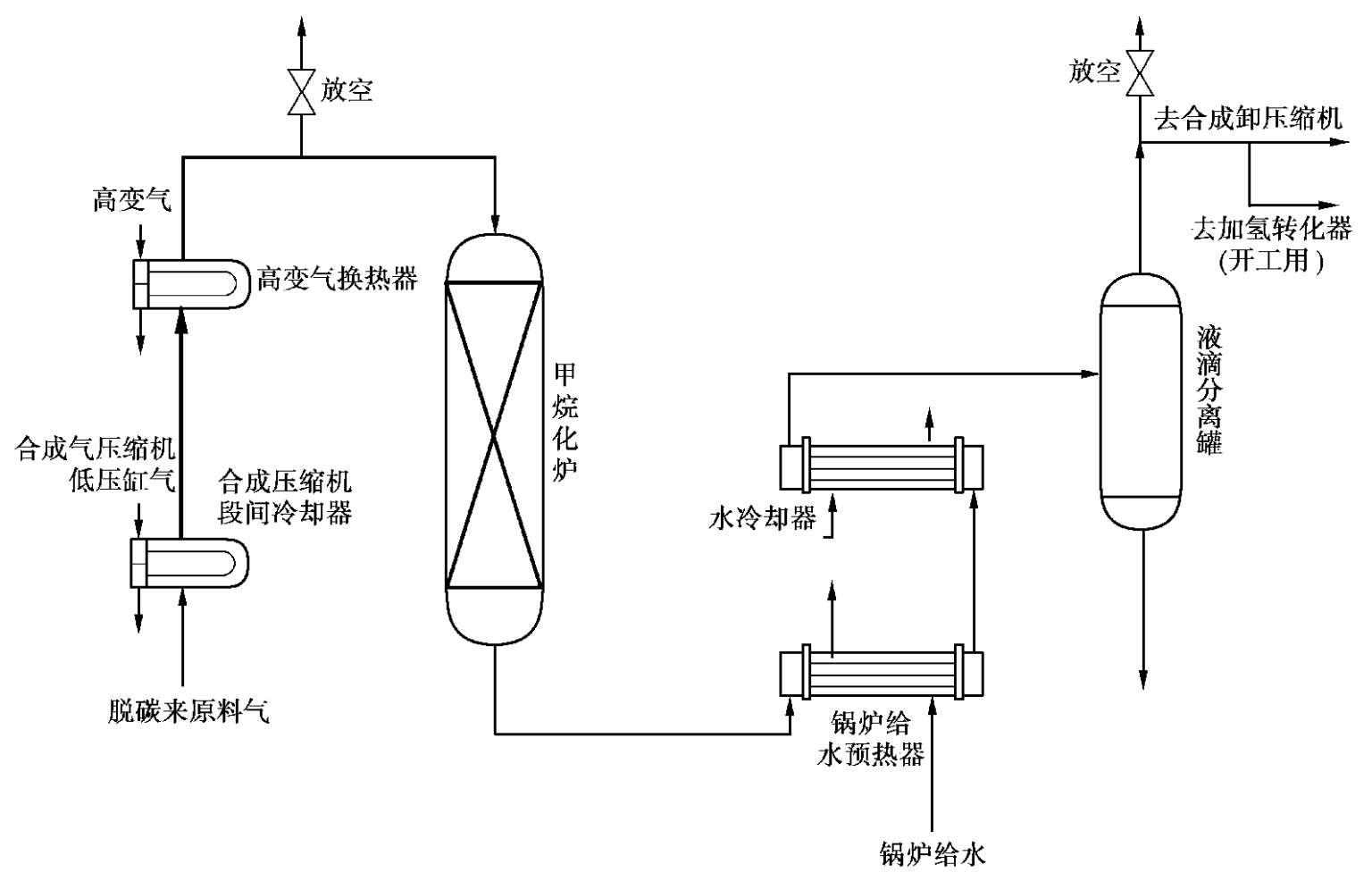
图 1.28 甲烷化法精制原料气流程
从CO 2 吸收塔来的气体,进入合成压缩机低压缸出口换热器,并预热到所需温度,然后进入高变换热器,加热到反应所需的温度,进入甲烷化炉。反应后的气体温度升高后,首先进入锅炉给水预热器降温,然后进入水冷却器冷却。气体进入液滴分离罐,分出水分后送往合成气压缩机。