
下载掌阅APP,畅读海量书库
立即打开




美国有关药品监管的法律法规分为三个层次。
第一个层次是联邦法律,美国宪法将所有的立法权授予众议院和参议院两院组成的国会(Congress)。当法案由两院的成员提出后,经过相关主题委员会审查或召开听证会,由两院投票通过,最后交由总统签署。签署后的法案则视为生效。正式发布的成文法会根据其规范的内容,被编入《美国法典》(United States Code,U.S.C.)中54个相应主题的相关卷中,其中主题21包含了有关食品与药品的成文法。《联邦食品、药品和化妆品法令》在《美国法典》主题21中321~399i部分中可以找到,其中351~360fff-7部分是有关药品和医疗器械的
 。另外有关规范生物制品的是《公众健康服务法案》在《美国法典》主题42中可以找到
。这两部法律再加上其他一些联邦法规组成FDA遵从的法规框架,如《处方药付费法案》(Prescription Drug User Fee Act,PDUFA)、《FDA现代化法案》(Food and Drug Administration Modernization Act,FDAMA)、《FDA现代化法案修正案》《FDA安全与创新法案》(Food and Drug Administration Safety and Innovation Act,FDASIA)等。
。另外有关规范生物制品的是《公众健康服务法案》在《美国法典》主题42中可以找到
。这两部法律再加上其他一些联邦法规组成FDA遵从的法规框架,如《处方药付费法案》(Prescription Drug User Fee Act,PDUFA)、《FDA现代化法案》(Food and Drug Administration Modernization Act,FDAMA)、《FDA现代化法案修正案》《FDA安全与创新法案》(Food and Drug Administration Safety and Innovation Act,FDASIA)等。
第二个层次是FDA在以上法律基础上依照《行政程序法案》或其他联邦法律制定的一系列法规,FDA可以通过法规来落实法律的实施。法规的作用是进一步解释和描述法律的强制性执行。起草法规过程中包括“告知和征求意见”,允许公众对于FDA提出的法规在最终版发布前提出意见。FDA颁布的法规也属于联邦法律。新法规或现有法规的变更内容,会在联邦公报(Federal Register)上进行公布。规范食品与药品的法规被收录在《联邦法规汇编》(Code of Federal Regulations,CFR)的主题21
中。规范药品的联邦法规主要有以下几种。
(1)新药和仿制药申请要求(主题21中的314~600部分)。
(2)药品生产、包装或储存的良好管理规范(主题21中的210部分)。
(3)药品制剂的良好管理规范(主题21中的211部分)。
第三个层次是FDA根据“良好指南制定规范”发布的FDA指南,指南说明了FDA对于监管内容的目前的想法。指南通常不具有法律效力。FDA编制的指南涵盖了所有药品生命周期的各个阶段,包括临床试验、药学研究、质量合规等 [1] 。
美国食品药品监督管理局(FDA)是美国政府在健康与人类服务部(Department of Health and Human Services,DHHS)下属的执行机构,是保护公共健康和安全的科学法律机构,监管范围非常广,主要包括食品、药品、生物制品、医疗器械、放射性电子产品、烟草等与公众健康安全息息相关的各行各业。通过保证人用药品、兽药、生物制品和医疗器械的安全和有效以及食品供应、化妆品和放射产品的安全进而保护公众健康,帮助加快创新,使药品和食品更有效、安全、易获得,保证公众得到所使用的药品和食品的正确、科学的信息。
FDA总部位于美国马里兰州,在全国范围有5个区域办公室,20个地区办公室,在中国、印度等地设有海外办公室。
美国FDA由局长办公室、四大监管核心职能的部门,包括药品和烟草、食品和兽药、全球监管运营和政策以及日常运营。图2-1为FDA的组织架构图。
药品与烟草办公室领导药品、生物制品、医疗器械和烟草产品各中心,并负责跨部门的高层协调。其中药品审评和研究中心及生物制品审评和研究中心主要负责药品和生物制品的新药以及新适应证的审评和批准。
药品审评和评价中心通过确保人们获得安全、有效的药品来促进基本的公众健康。CDER负责监管非处方药和处方药,包括治疗性生物制品和仿制药。主要包括以下与药品直接相关的办公室。
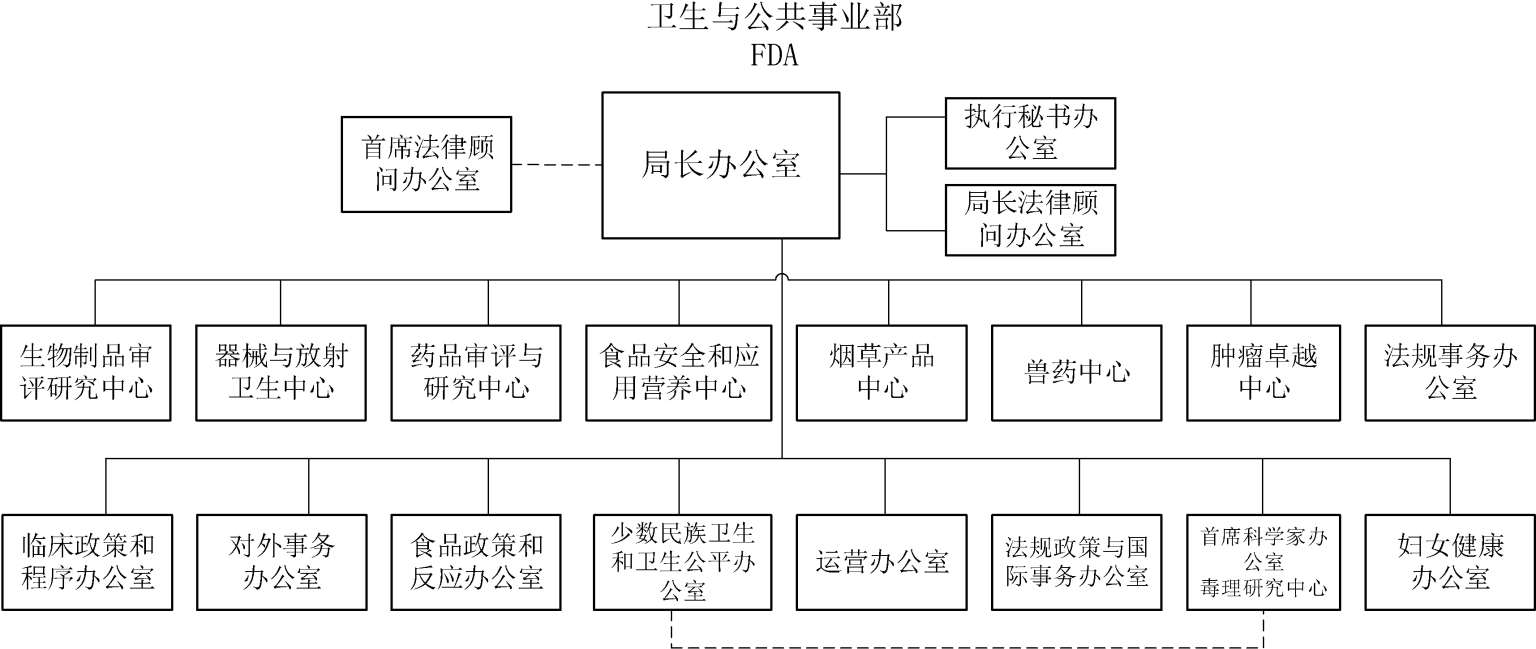
图2-1 美国FDA组织架构图
(1)新药办公室(OND)
新药办公室包括6大办公室和19个部门。主要负责审评并批准药品申请,为确保有效的审评流程而制定指南和方针。OND审评药品申请,与工业界互动,并对药品是否收益大于风险作出最终决定。
(2)仿制药办公室(OGD)
通过法规流程保证美国公众获得安全、有效和高质量的仿制药。FDA批准的仿制药占美国药品处方量的88%。
(3)药物质量管理办公室(OPQ)
药物质量管理办公室的承诺是为美国公众提供质量符合要求的药品,它整合了药品的审评、检查、上市监管、政策和研究活动以在全球范围内加强药品质量。OPQ提倡“一个质量声音”,在所有生产场地(国内和国外)以及所有药品领域包括新药、新生物制品、仿制药、生物类似药以及非处方药和调剂的药品,创造统一的质量程序。OPQ也鼓励采用新技术来提高药品质量和潜在的重振药品制造业。
(4)合规办公室(OC)
合规办公室通过合规政策和基于风险的强制法规行动使病人免于接受质量差、不安全和疗效差的药品。根据法律和科学制订战略和基于风险的决定以强化全球合作,推进自愿合规和采取果断、快速的行动以保护病人。负责通过实施工厂检查、产品测试和其他上市前及上市后的合规行动来监控药品质量,为全球FDA办公室采取调查和法规行动提供支持和指导。警告信通常由该办公室签署发出。
生物制品审评和评价中心的承诺是通过监管生物制品及相关产品包括血液、疫苗、抗过敏产品及组织、细胞和基因治疗产品来保证和加强公众健康。CBER负责通过评估制造商提交的科学的临床数据来决定新的生物制品以及已批准生物制品新适应证是否符合CBER的批准标准。CBER针对预期使用人群和产品的预期用途的风险—效益比做出决定。CBDR的授权来自“公众健康服务法案”和“食品、药品和化妆品法案”的一些部分。
监管事务办公室隶属于全球监管运营和政策办公室,是所有区域办公室的领导办公室,ORA检查监管的产品及其制造商,进行采样分析,评估进口产品,对客户投诉、紧急情况或犯罪行为进行调查。ORA也与各级地区及境内外的类似机构一起合作。
FDA的药品生产质量监管主要有3组机构:①审评部门,是位于CDER和CBER内部的负责新药和仿制药审评的机构;②合规部门,合规办公室负责药品生产质量管理规范法规和相关指南的制定,向ORA提出现场检查要求,审评检查报告并作出合规决定;③检查部门,ORA负责GMP现场检查及生物制品、医疗器械和食品的现场检查以及实验室检测工作。
CGMP是指FDA颁布的现行的GMP法规,FDA通过监控药品制造商是否符合现行GMP法规来保证药品的质量。现行药品GMP包含了方法、厂房和药品生产、工艺和包装的控制的最低要求,法规确保了药品可以安全使用并包含了说明书或标签上申明的成分和组成。CGMP的要求比较灵活,允许药品制造商自行决定通过科学良好的设计、工艺方法和测试程序来最好地实施必要的控制。法规的灵活性还允许制造商使用现代技术和创新方法来持续改进,寻求高质量。因此,“C”在CGMP中代表“现行的”,要求制造商使用最新的技术和系统以满足法规要求,防止污染、混淆和出错。采用10年或20年前的技术是不合适的。美国FDA人用药品GMP由两部分组成,即CFR Part 210
(药品生产、包装或储存的良好管理规范,总则),和CFR Part 211
(药品制剂的当前生产质量管理规范)。
FDA的合规程序指导FDA工作人员评估行业是否符合食品、药品、化妆品以及其他法律的活动。合规程序指南手册根据不同产品进行分类,相关内容颁布于FDA网站
。
现场检查可分为新药和仿制药申请的批准前检查、常规GMP检查(通常每两年一次)及有因检查。其中,批准前检查是应审评部门要求,由合规部门发起、检查部门实施的,审评和合规人员有时也会参与;常规GMP检查由合规部门通过风险评估的模型建议检查部门执行。检查部门也可发起常规GMP检查和有因检查。
检查的内容:基于风险和系统(即质量系统、设施和设备系统、物料控制系统、生产系统、包装和贴签系统、实验室控制系统等GMP六大系统),分为全面检查(至少查4个系统)和简略检查(至少查2个系统),其中质量系统是必查的,其他系统由检查部门地区办公室根据风险评估选择。
检查的时间:批准前检查中,原料药一般1周,无菌制剂最长,一般2周;常规检查可能更长;有因检查视事件性质而定。FDA现场检查结束后,检查员初步评估GMP合规/违规性,在离开工厂前向工厂经营者或代理人提供GMP违规的通知,即“483”缺陷表。“483”缺陷表是检查员填写违规发现表格的名称,全称为“FDA表483现场检查观察”。检查员在检查结束回到办公室后,在两周内完成正式检查报告(Establishment Inspection Report,EIR),如有需要提交推荐的监管方案,交由上级官员评估。
如果出现违规即被视为一个“案件(Case)”,每个“案件”都必须通过监管措施解决。合规审评员负责审评“案件”解决方案,并批准解决“案件”相应的整套监管方案。FDA法律顾问负责审评由CDER批准的“案件”整套监管方案在法律和证据方面的完整性和一致性。由地区办公室负责监督参与“案件”整套监管方案执行的工厂管理层或个人。当工厂的整改完成并符合要求时,经合规部门同意,工厂的合规状态可以恢复正常,回到FDA正常(非“案件”性)监管程序中。
[1] 所有与药品相关的指南可以参见网址https://www.fda.gov/drugs/guidances-drugs/all-guidances-drugs,大约810个。
所有与生物制品相关的指南可以参见网址https://www.fda.gov/vaccines-blood-biologics/guidance-compliance regulatory-information-biologics/biologics-guidances,大约541个。