
下载掌阅APP,畅读海量书库
立即打开



重量分析法是根据生成物的重量来确定被测组分含量的方法。在重量分析中,一般是先使被测组分从试样中分离出来,转化为可准确称量的形式,然后用称量的方法测定该组分的含量。
1.分类 根据被测组分与试样中其他组分分离所采用方法的不同进行分类,重量分析法分为挥发法、萃取法、电解法、沉淀法。
2.特点
(1)直接称量得到分析结果,不用基准物质比较,准确度高。
(2)操作繁琐,程序长,费时多。
1.直接法 利用加热或其他方法使样品中的被测组分气化逸出。然后用吸收剂将气化组分全部吸收,通过称量吸收剂的增重来确定被测成分的含量的方法。
如试样中CO 2 的测定,以碱石灰为吸收剂。
2.间接法 利用加热或其他方法使样品中的被测组分气化逸出后,称量其残渣,由样品的减量来确定该挥发组分的含量。
如测定湿存水或结晶水,加热烘干至恒重,试样减轻的质量或用干燥剂吸收水蒸汽后增加的质量来确定水的质量。
萃取法是利用萃取剂把被测组分萃取出来,蒸发除去萃取剂,称出萃取物的质量,从而确定被测组分含量的方法,又称提取法。
如测定粮食中的脂肪含量,可用乙醚在一定条件下将脂肪提取到乙醚中,然后,将乙醚蒸发,由容器在蒸发前后质量之差,可求脂肪含量。
电解法是利用电解原理,用电子作沉淀剂使金属离子在电极上还原析出,然后称量,求得其含量。
如测溶液Cu 2+ 含量,可通过电解使试液中的Cu 2+ 在阴极上析出,由电解前后阴极质量之差计算Cu 2+ 的含量。
(一)概述
1.沉淀法 是利用沉淀反应将被测组分以难溶化合物的形式从溶液中分离出来,然后经过滤、干燥、烘干或灼烧后,得到有固定组成的称量形式,称重后计算其含量的方法。
2.对沉淀形式的要求 沉淀的化学组成称沉淀形式。
(1)沉淀的溶解度要足够小。沉淀溶解损失的量应不超出分析天平的称量误差范围。
(2)沉淀必须纯净,尽量避免混入杂质。
(3)沉淀应易于过滤和洗涤。尽量获得大颗粒沉淀。
(4)沉淀应易于转化为称量形式。
3.对称量形式的要求 沉淀经干燥或灼烧处理后,供最后称量的化学组成称称量形式。
(1)称量形式必须有确定的化学组成。
(2)称量形式要稳定。
(3)称量形式的式量要大,以减小称量误差,提高分析结果的准确度。
(二)沉淀的形态与沉淀的形成
1.沉淀的形态 沉淀按照其物理性质不同,可粗略地分成晶型沉淀和无定形沉淀(非晶型沉淀或胶状沉淀)。
晶形沉淀:颗粒大,内部排列较规则,结构紧密。
无定性沉淀:沉淀颗粒小,由结构疏松聚集在一起的微小沉淀颗粒组成,呈疏松的絮状沉淀,整个沉淀体积庞大。
在重量法中,希望得到的晶形沉淀颗粒大,无定形沉淀紧密。
2.沉淀的形成 在待测定离子的溶液中加入沉淀剂,当溶液中构晶离子的浓度的乘积超过溶度积时,就有可能生成沉淀。
晶核形成过程有两种成核作用。构晶离子在过饱和溶液中聚集、自发形成晶核称均相成核作用;溶液中不可避免地混有不同数量的固体微粒(试剂、溶剂、灰尘都会引入杂质),它们的存在可起到“晶种”作用,“诱导”构晶离子在“晶种”上沉积形成晶核,这种成核作用称异相成核作用。
晶核形成后,溶液中的构晶离子在晶核上沉积并逐渐长大成肉眼仍看不见的沉淀微粒。这种微粒有聚集长大的两种可能趋势。一种是构晶离子继续按一定的晶格定向有序的排列,成长为大颗粒晶形沉淀;另一种是沉淀微粒来不及继续定向排列就以较快速度无序聚集长大形成无定形沉淀。这两种不同趋势主要取决于成核速度、聚集速度和定向速度的相对大小。而这些因素既与沉淀物质的本性有关,更与沉淀条件有关。
溶液的过饱和程度直接影响晶核的生成速度和沉淀颗粒的大小。
当溶液的相对过饱和度较小时,沉淀生成的初速度很慢,此时异相成核是主要的成核过程;由于溶液中外来固体颗粒的数目是有限的,构晶离子只能在这有限的晶核上沉积长大,从而有可能得到较大的沉淀颗粒。
当溶液的相对过饱和度较大时,由于沉淀生成的初速度较快,大量构晶离子必然自发地生成新的晶核,而使均相成核作用突显起来,溶液中晶核总数也随着相对过饱和度的增大而急剧增大,致使沉淀的颗粒减小。
一般地,极性较强的盐类如BaSO 4 等,一般具有较大的定向速度,故常生成晶形沉淀。
氢氧化物,特别是高价金属离子的氢氧化物,它们的溶解度很小,沉淀时溶液的相对过饱和度较大;又由于含有大量的水分子而阻碍着离子的定向排列,因此定向速度较小,一般易生成体积庞大、结构疏松的无定形沉淀。
沉淀的类型不仅取决于沉淀物质的本性,也与进行沉淀的条件密切相关,成核过程和晶体的成长过程都对沉淀颗粒的大小有影响。
(三)沉淀完全的程度与影响因素
利用沉淀反应进行重量分析时,误差的主要来源是沉淀的溶解损失,所以要求沉淀反应尽可能地进行完全。沉淀是否完全,取决于沉淀的溶解度的大小。溶解度越小,沉淀越完全;溶解度越大,沉淀进行越不完全。
重量法要求沉淀的溶解损失应不超过分析天平的称量绝对误差(±0.2mg)。
1.溶度积与溶解度
(1)溶解度沉淀MA在水中的溶解,有两步平衡。
MA(固)=MA(水)= M + + A -
根据MA(固)和MA(水)之间的沉淀平衡可得:
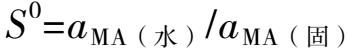
考虑到固体活度a MA(固) =1,溶液中中性分子的活度系数近似为1,则:
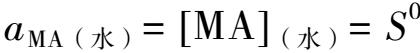
S 0 表示MA(水)的溶解度,即MA在水溶液中以分子状态或离子对状态存在时的活度,称为分子溶解度。
(2)溶度积活度积常数:K ap =a M+ ·a A-
K sp =[M + ][A - ]
K ap 称为活度积,它仅与温度有关。
K sp 称为溶度积,它除了受温度影响外,还与溶液的离子强度有关。
在重量分析中由于难溶化合物的溶解度一般较小,溶液中的离子强度不大,通常可以忽略离子强度的影响,可以用活度积K ap 代替溶度积K sp 。
2.影响沉淀溶解度的因素
(1)同离子效应当沉淀反应达到平衡后,如果向溶液中加入含有某一构晶离子试剂或溶液,可降低沉淀的溶解度。但沉淀剂加得太多会引起盐效应、酸效应及络合效应等副反应,反而使溶解度增大。所以沉淀剂一般过量50%~100%;对于灼烧时不易挥发除去的沉淀剂,则一般以过量20%~30%为宜。
(2)配位效应当溶液中存在能与沉淀的构晶离子形成配合物的配位剂时,则沉淀平衡向沉淀溶解的方向移动,使沉淀的溶解度增大。这种现象称为配位效应。
配位效应对沉淀溶解度的影响,与配位剂的浓度及生成的配合物的稳定性有关。配位剂的浓度越大,生成的配合物越稳定,沉淀的溶解度越大。
(3)水解作用有些构晶离子能发生水解作用,使沉淀溶解。
(4)形成胶体溶液胶体微粒小,过滤时,易透过滤纸引起损失。常加入适量电介质以防止胶溶作用。
(5)其他影响因素①温度:温度升高使溶解度增大。②溶剂:大多数无机物沉淀在有机溶液中的溶解度比在纯水中小。③形成胶体溶液:过滤时极易穿透滤纸而引起损失,加热或加入大量电解质可破坏胶体。④颗粒大小:颗粒越小,溶解度越大。⑤沉淀析出形态的影响:亚稳态>稳态。
(四)沉淀的纯度
在实际操作中,当沉淀从溶液中析出时,或多或少地会夹杂溶液中的其他组分,使沉淀不纯,其中主要有两种干扰方式,即共沉淀和后沉淀。
1.共沉淀 产生共沉淀的原因主要有表面吸附、形成混晶和吸留。
(1)表面吸附共沉淀表面有剩余电荷,吸附构晶离子,形成吸附层,再吸附相反电荷离子(称抗衡离子),形成扩散层,组成了双电层。
1)吸附规律:
第一吸附层:优先吸附构晶离子。
第二吸附层:①与构晶离子形成溶解度、离解度最小的化合物。②离子浓度越大,越易被吸附。③离子电荷高的优先吸附。
2)影响因素:①颗粒小,比表面积大,吸附杂质多。②吸附是放热过程,温度升高,吸附杂质量减少。
3)防止:洗涤。
(2)混晶或固溶体与构晶离子半径相近,形成的晶体结构相近的离子代替构晶离子形成混晶,不能洗去,应在沉淀前进行分离。
(3)吸留和包夹表面吸附的抗衡离子在沉淀生长时被覆盖包藏在里面不能洗去,陈化可减小,严重时,需重沉淀。
2.后沉淀 当沉淀析出后,与母液一起放置的过程中,溶液中的杂质离子会慢慢地在沉淀表面上沉积,这种现象称为后沉淀。
避免或减少继沉淀的主要方法是缩短沉淀和母液共置时间。
3.提高沉淀纯度的措施 为减少因共沉淀和后沉淀造成的影响,可以采取以下措施:
(1)选择适当的沉淀步骤。测少量组分含量时,首先沉淀含量少的组分。
(2)降低易被吸附的杂质离子的浓度。必要时应预先分离除去或掩蔽。
(3)选择适当的沉淀剂。选用有机沉淀剂可有效减少共沉淀。
(4)再沉淀。有效减小吸留或包埋的共沉淀及后沉淀现象。
(五)沉淀的条件
1.晶形沉淀的沉淀条件 对于晶型沉淀,主要考虑如何得到较大的沉淀颗粒,以使沉淀纯净并易于过滤、洗涤。一般采用的沉淀条件是:稀、热、慢、搅、陈。
(1)沉淀反应应在适当稀的溶液中进行。
(2)在热溶液中进行沉淀。
(3)在不断搅拌下缓慢滴加沉淀剂。
(4)沉淀时应不断搅拌。
(5)陈化。
2.无定性沉淀的沉淀条件 对无定性沉淀主要考虑如何加快沉淀微粒的凝聚,使沉淀紧密,减少对杂质的吸附。采用的沉淀条件是:浓、热、电、不陈。
(1)沉淀反应应在比较浓的溶液中进行,而且要较快地加入沉淀剂。
(2)在热溶液中进行沉淀。
(3)加入大量的电解质,防止沉淀的胶溶。
(4)不需陈化。
(六)称量形式与结果计算
1.换算因数F 根据称量形式与被测组分表示式之间的化学计量关系求得。
F=(待测组分的摩尔质量×a)/(称量形式的摩尔质量×b)
a、b:使分子和分母中所含主体元素的原子个数相等时需乘以的系数。
2.被测组分的质量分数 被测组分%=(称量形式的质量×换算因数)/试样质量。
(一)萃取
溶质从一种溶剂转移到另一种溶剂的过程称为萃取。
1.萃取的基本原理 萃取是利用物质在两种不互溶(或微溶)溶剂中溶解度或分配比的不同来达到分离、提取或纯化物质的操作。
分配定律是液-液萃取的主要理论依据。在两种互不相溶的混合溶剂中加入某种可溶性物质时,它能以不同的溶解度分别溶解于此两种溶剂中。实验证明,在一定温度下,若该物质的分子在此两种溶剂中不发生分解、解离、缔合和溶剂化等作用,则此物质在两液相中浓度之比是一个常数,假如一种物质在两液相A和B中的浓度分别为c A 和c B ,则在一定温度下,c A /c B =K,K是一常数,称为分配系数。K可以近似地看作此物质在两溶剂中溶解度之比。
由于多数有机物在有机溶剂中有更好的溶解性,因此用有机溶剂来萃取水溶液中的有机物是萃取的典型实例。从水溶液中萃取有机物时,选择合适萃取溶剂的一般原则是要求溶剂在水中溶解度很小或几乎不溶;被萃取物在溶剂中要比在水中溶解度大;溶剂与水和被萃取物都不反应;溶剂的沸点不易过高;萃取后溶剂易于和溶质分离开。因此,最好用低沸点的溶剂,萃取后溶剂可用常压蒸馏回收。此外还要兼顾溶剂价格、毒性、安全等因素。
经常使用的溶剂有乙醚、二氯甲烷、石油醚、四氯化碳、氯仿、乙酸乙酯和苯等。一般水溶性较小的物质可用石油醚萃取,水溶性较大的可用乙醚萃取,水溶性极大的可用乙酸乙酯萃取。也可以用水来萃取有机溶剂以除去不要的组分,这个过程常称为洗涤。如果有机层中含有需要除去的酸(如盐酸),就可用水或稀的弱碱水溶液来洗涤。如果要除去碱性物质,可用稀酸水溶液洗涤。
用一定量的有机溶剂萃取时,把溶剂量分成多次萃取比用全部量做一次萃取效果要好。例如在100ml水中溶有4g丁酸,15℃时用100ml苯来萃取其中的丁酸,用100ml苯一次萃取时,在水中丁酸的剩余量为1.0g,但若将100ml苯分3次萃取,则剩余量减少为0.5g(此数值可由公式计算得出)。一般萃取次数为3~5次即可。
另外,在萃取时,若在水溶液中加入一定量的电解质(如氯化钠),利用“盐析效应”以降低有机物和萃取溶剂在水溶液中的溶解度,可提高萃取效率。如用乙醚萃取水层时,常用饱和氯化钠水溶液代替水来洗涤醚层。
2.分液漏斗的使用 在实验室中进行液-液萃取时,一般在分液漏斗中进行。
分液漏斗内加入的液体量不能超过容积的3/4。为防止杂质落入漏斗内,应盖上漏斗口上的塞子。放液时,磨口塞子上的凹槽与漏斗口颈上的小孔要对准,这时漏斗内外的空气相通,压强相等,漏斗里的液体才能顺利流出。当分液漏斗中的液体向下流时,活塞可控制液体的流量,若要停止分液,就要将活塞紧紧关闭。
分液漏斗也可用于分离互不相溶的液体。使用分液漏斗分液时根据“下流上倒”的原理,打开活塞让下层液体全部流出,然后关闭活塞,上层从上口倒出。分液漏斗不能加热,漏斗用后要洗涤干净。长时间不用的分液漏斗要把旋塞处擦拭干净,旋塞与塞槽之间放一纸条,以防磨砂处粘连。
(1)使用前的准备工作
1)分液漏斗上口的顶塞应用小线系在漏斗上口的颈部,旋塞则用橡皮筋绑好,以避免脱落打破。
2)取下旋塞并用纸将旋塞及旋塞腔擦干,在旋塞孔的两侧涂上一层薄薄的凡士林,再小心塞上旋塞并来回旋转数次,使凡士林均匀分布并透明。但上口的顶塞不能涂凡士林。
3)使用前应先用水检查顶塞、旋塞是否紧密。倒置或旋转旋塞时都必须不漏水,方可进行使用。
(2)萃取与洗涤操作把分液漏斗放置在固定于铁架台的铁环(用石棉绳缠扎)上。关闭旋塞并在漏斗颈下面放一个锥形瓶。由分液漏斗上口倒入溶液与溶剂(液体总体积应不超过漏斗容积的2/3),然后盖紧顶塞并封闭气孔。取下分液漏斗,振摇使两层液体充分接触。振摇时,右手捏住漏斗上口颈部,并用食指根部(或手掌)顶住顶塞,以防顶塞松开。用左手大拇指、食指按住处于上方的旋塞把手,既要能防止振摇时旋塞转动或脱落,又要便于灵活地旋开旋塞。漏斗颈向上倾斜30°~45°角。
(3)两相液体的分离操作分液漏斗进行液体分离时,必须放置在铁环上静置分层;待两层液体界面清晰时,先将顶塞的凹缝与分液漏斗上口颈部的小孔对好(与大气相通),再把分液漏斗下端靠在接受瓶壁上,然后缓缓旋开旋塞,放出下层液体,放时先快后慢;当两液面界限接近旋塞时,关闭旋塞并手持漏斗颈稍加振摇,使黏附在漏斗壁上的液体下沉,再静置片刻;下层液体常略有增多,再将下层液体仔细放出;此种操作可重复2~3次,以便把下层液体分净。当最后一滴下层液体刚刚通过旋塞孔时,关闭旋塞。待颈部液体流完后,将上层液体从上口倒出。绝不可由旋塞放出上层液体,以免被残留在漏斗颈的下层液体所沾污。
不论萃取还是洗涤,上下两层液体都要保留至实验完毕。否则一旦中间操作失误,就无法补救和检查。
分液漏斗与碱性溶液接触后,必须用水冲洗干净。不用时,顶塞、旋塞应用薄纸条夹好,以防粘住(若已粘住,不要硬扭,可用水泡开)。当分液漏斗需放入烘箱中干燥时,应先卸下顶塞与旋塞,上面的凡士林必须用纸擦净,否则凡士林在烘箱中炭化后,很难洗去。
3.溶液中物质的萃取 首先,应选择容积较液体体积大一倍以上的分液漏斗,把活塞擦干,在离活塞孔稍远的两端薄薄地涂上一层凡士林,塞好活塞并旋转几圈,使凡士林分布均匀,但活塞孔附近及上口的塞子不能涂凡士林,以免污染萃取液。使用前先用水检查上口塞子和活塞是否密封,确认不漏水时方可使用。
然后,将漏斗放在固定于铁架台上的铁圈中,关好活塞,将要萃取的水溶液和其1/3体积的萃取剂自上口倒入漏斗中,塞紧塞子。
取下分液漏斗,用手掌顶住漏斗上口的塞子并握住漏斗,另一只手握住漏斗活塞处,大拇指压紧活塞,将漏斗倒置,活塞端向上呈45°角握稳,打开活塞,先将漏斗活塞端朝向无人处放气一次,然后关闭活塞将漏斗上下振摇几次,放气,如此反复几次。在开始时,振摇要慢,漏斗振摇后内部的压力通常很大,如振摇后乙醚可产生40~66.7kPa的蒸气压,加上原来空气和水蒸汽的压力,漏斗中的压力就大大超过了大气压。如果不及时放气,塞子就可能被顶开而损失产品。
经过几次振摇后,将漏斗放到铁圈中静置,上口的活塞应对准出气口,如果分液漏斗没有出气口应将上口塞子松动后再放置,以使内部的压力及时排除。
待两层液体完全分开后,打开上面的塞子,再将活塞缓缓旋开,下层液体自活塞放出。分液时一定要尽可能分离干净,中间层视具体情况决定放入下层或留在上层,漏斗下口的细管中经常会残留一部分液体,应轻轻震荡漏斗,使其流出。然后将上层液体从分液漏斗的上口倒出。
如此反复3~5次,将所有的萃取液合并即完成萃取操作。
很多有机溶剂是无色液体,当溶解了溶质以后密度会发生变化,因此实际操作中要经常判断哪一层是水层。可以取其中一层的少量液体,置于试管中,滴加少量水来鉴别。
在萃取操作中,会经常产生乳化现象。乳化现象的产生原因比较复杂,有时是由碱性物质引起,有时是两相的相对密度相近所引起,还有的乳化是由被萃取物引起。一旦出现乳化现象,两相分离就很难进行,必须先破除乳化,用来破坏乳化的常用方法有:
①较长时间静置。②酸化,若因溶液碱性而产生乳化,常可加入少量稀酸破除乳化。③加入破乳剂,如乙醇、磺化蓖麻油等。④利用盐析作用,若因两种溶剂能部分互溶而发生乳化,可以加入少量电解质,利用盐析作用加以破坏。在两相相对密度相差很小时,加入氯化钠,也可以增加水相的相对密度。
萃取操作注意事项:
①分液漏斗中的溶液不可太满,否则振摇时不能使溶液和溶剂充分接触。
②将漏斗活塞关严后,再装溶液。以免溶液流失,影响产率。另外,溶液倒入前,或在静置过程中,应在漏斗下方,置一锥形瓶作为接受瓶,以备出错时补救。
③振摇时,应及时排气。以免分液漏斗内压过高,溶液从玻璃接缝中渗出。
④振摇后,静置要充分。分层完全后再分离。
⑤溶液分离时,要按照下层液体由下部支管放出,上层液体应由上口倒出的原则。
⑥萃取或洗涤过程中,上下两层液体都应保留至实验完毕时,否则,如果中间的操作失误,便无法补救。
⑦要弄清哪一层是水溶液。若搞不清,可任取一层的少量液体,置于试管中,并滴少量自来水,若分为两层,说明该液体为有机相,若加水后不分层则是水溶液。
⑧在萃取时,在水溶液中加入一定量的电解质(如氯化钠),以降低有机物在水中的溶解度,提高萃取效果。
(二)沉淀的过滤和洗涤
需要灼烧的沉淀用滤纸过滤,准备烘干的沉淀用玻璃砂芯滤器过滤。
采用倾泻法过滤。
1.滤纸的选择 滤纸是最常用的过滤介质,按过滤速度(或分离性能)不同,滤纸可分为快速、中速和慢速三种,可根据沉淀的性质和漏斗的规格大小来选用。例如,晶型沉淀(BaSO 4 、CaC 2 O 4 等)可选用直径9~11cm、慢速的定量滤纸,而对于胶状沉淀(Fe 2 O 3 ·xH 2 O等),则应选用直径为11~12.5cm,快速的定量滤纸。
另外,由于滤纸具有强的吸水性,不能将沉淀经滤纸过滤后直接进行干燥再称重。一般总是将沉淀过滤后,将滤纸灰化。定量分析用的滤纸称为无灰滤纸,在制造这种滤纸时已用盐酸和氢氟酸除去其中的杂质。一张定量滤纸的质量约为1g,其灰份含量小于0.1mg。
滤纸大小应根据沉淀量的多少而定,沉淀体积应低于滤纸容积的1/3。此外还要和漏斗相适应,一般滤纸放入后,其边缘应低于漏斗口0.5~1.0cm。
2.滤纸的折叠与安放 滤纸的折叠:一般将滤纸对折,然后再对折(暂不要折固定)成四分之一圆,放入清洁干燥的漏斗中,如滤纸边缘与漏斗不十分密合,可稍稍改变折叠角度,直至与漏斗密合,再轻按使滤纸第二次的折边固定,取出成圆锥体的滤纸,把三层厚的外层撕下一角,以便使滤纸紧贴漏斗壁(图5-1),撕下的纸角保留备用。若用布氏漏斗,则要选择与漏斗直径相适合的滤纸,而不需折叠。
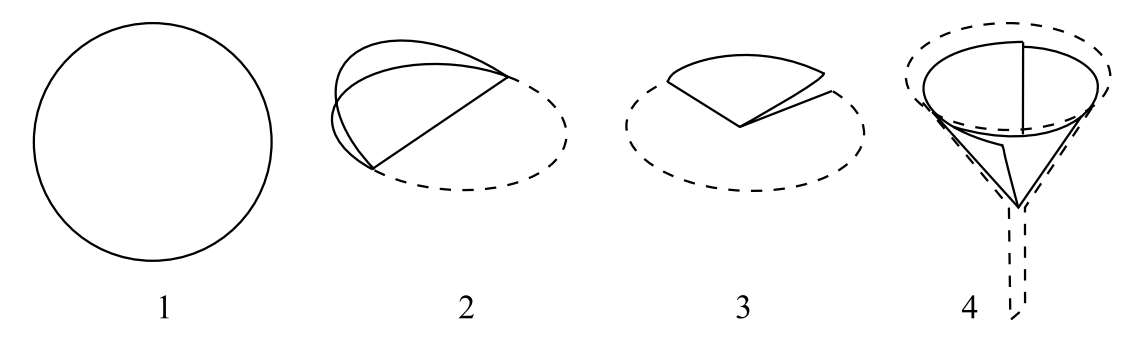
图5-1 滤纸的折叠和安放
滤纸的安放:把折好的滤纸放入漏斗,三层的一边应对应漏斗出口短的一边。用食指按紧,用洗瓶吹入水流将滤纸湿润,轻按压滤纸边缘使锥体上部与漏斗密合,但下部留有缝隙,加水至滤纸边缘,此时空隙应全部被水充满,形成水柱,放在漏斗架上备用。
为加快过滤速度也可采用下面的方法折叠滤纸和配套漏斗(图5-2):
图5-2 滤纸的其他折叠方法
如图从(a)折到(c)将已折成半圆形的滤纸分成八个等份,再如(d)将每份的中线处来回对折(注意折痕不要集中在顶端的一个点上,以免将滤纸弄破)。
3.沉淀的过滤和洗涤 一般采用“倾注法”过滤:即先把沉淀上层的清液(注意不要搅动沉淀)沿玻璃棒倾入漏斗,令沉淀尽量留在烧杯内。注意玻璃棒应垂直立于滤纸三层部分的上方,尽量接近而不接触滤纸,倾入的溶液面应不超过滤纸边缘下5~6mm处,漏斗颈下端不应接触溶液。
“一贴二低三靠”:滤纸背面紧贴漏斗壁,用水打湿不出气泡为止,滤纸边缘低于漏斗;过滤时加入漏斗的溶液面低于滤纸边缘,过滤时,烧杯嘴与玻璃棒接触;玻璃棒与三层滤纸处相接触;漏斗嘴紧靠玻璃烧杯壁。
当暂停倾注时,应将烧杯沿玻璃棒慢慢上提同时缓缓扶正烧杯,待玻璃棒上的溶液流完后,把玻璃棒放回烧杯中但不可靠在烧杯嘴处。清液倾注完毕后,加适量洗涤液于烧杯中,待沉淀下沉后再倾注。洗涤液应少量多次加入,每次待滤纸内洗涤液流尽,再倾入下一次的洗涤液。
过滤时应观察滤液是否澄清,若发现混浊,则应将已过滤的部分,重新过滤。因此,用于承接滤液的器皿必须是干净的。
沉淀的转移:经多次倾注洗涤后,再加入少量洗涤液于烧杯中,搅起沉淀,使沉淀连洗涤液沿玻璃棒转移入漏斗的滤纸上,然后沾在烧杯壁的沉淀可用洗瓶吹洗并移入漏斗中。最后,用在准备滤纸所撕下的滤纸角擦净杯咀,玻璃棒,纸角一并置入漏斗。
沉淀的洗涤:沉淀全部转移后,继续用洗涤液洗涤沉淀,并使用适当检验方法检验沉淀是否洗涤干净(检验多余沉淀剂是否完全除去)。
4.抽滤 抽滤又称吸滤、减压过滤。利用抽气泵使抽滤瓶中的压强降低,由于瓶内与布氏漏斗液面上形成压力差,因而加快了过滤速度(图5-3)。
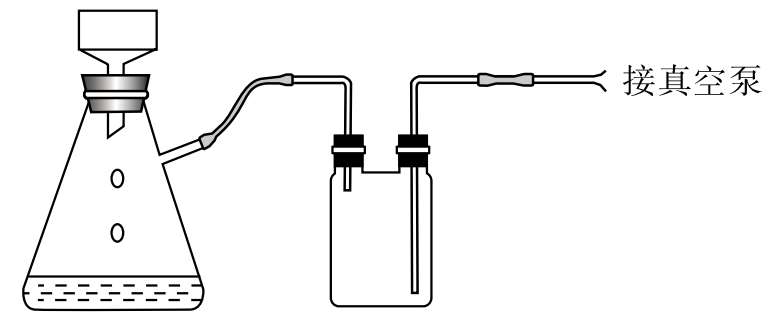
图5-3 抽滤装置示意图
过程:
①安装仪器,抽滤瓶上配一单孔塞,布氏漏斗安装在塞孔内。检查布式漏斗与抽滤瓶之间连接是否紧密,抽气泵连接口是否漏气。
②剪滤纸,使其略小于布氏漏斗,但要把所有的孔都覆盖住,并滴加蒸馏水使滤纸与漏斗连接紧密。
③将固液混合物转移到滤纸上。
④打开抽气泵开关,开始抽滤。
⑤过滤完之后,先抽掉抽滤瓶接管,后关抽气泵。
5.玻璃滤器过滤 对于烘干即可称重或热稳定性差的沉淀可用玻璃滤器过滤,这种漏斗无需用滤纸而可将沉淀或需分离的物质直接过滤在烧结玻璃片上,再在一定温度下烘至恒重即可(图5-4)。
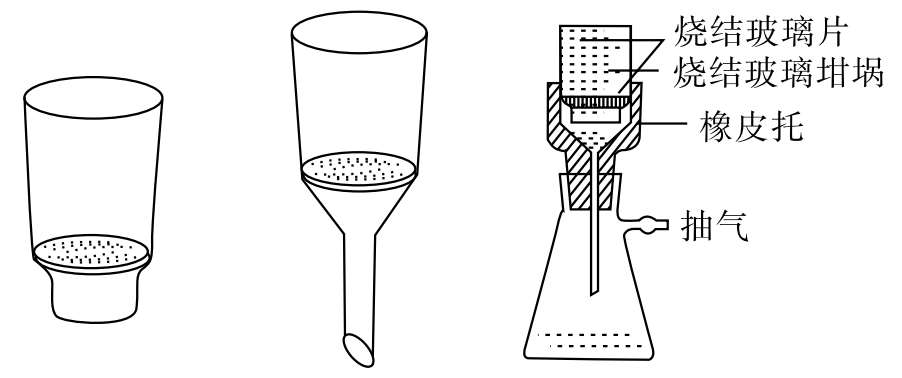
图5-4 玻璃滤器过滤装置示意图
(三)沉淀的烘干与灼烧
1.沉淀的包裹 用洁净的药铲或顶端扁圆的玻璃棒,将滤纸三层部分掀起两处,再用洁净的手指从翘起的滤纸下面将其取出,按图5-5打包。
2.坩埚的准备 坩埚用以盛载需要进行灼烧的沉淀。选择适当的坩埚,洗净,晾干并在灼烧沉淀的温度条件下经灼烧至恒重(即反复灼烧后其重量变化在0.2mg以内)。
包裹晶形沉淀的两种方法
图5-5 沉淀的包裹
胶状沉淀的包裹方法
3.将沉淀包转移入坩埚 当沉淀洗净,洗涤液已流干后,用玻璃棒将滤纸从三重厚的边缘开始将滤纸向内折卷,使滤纸圆锥体的敞口封上,成沉淀包,轻轻转动一下,把沉淀包取出,再将它倒置过来使尖端向上并放入坩埚中。这时,大部分的沉淀与坩埚底部接触,以便沉淀的干燥和灼烧。
4.沉淀的烘干和灼烧 将上述坩埚斜放在泥三角上,将坩埚盖半掩地倚在坩埚口(图5-6)。利用火焰将滤纸干燥、碳化,在这个过程中要适当调节火焰温度。当滤纸未干时,温度不宜过高以免坩埚破裂,在中间阶段将火焰放在坩埚盖之中心下方以便热空气反射入坩埚内部以加速滤纸干燥,随后将火焰移至坩埚底部提高火焰温度使滤纸焦化,最后适当转动坩埚位置,继续加热使滤纸灰化,灰化完全时沉淀应不带黑色。
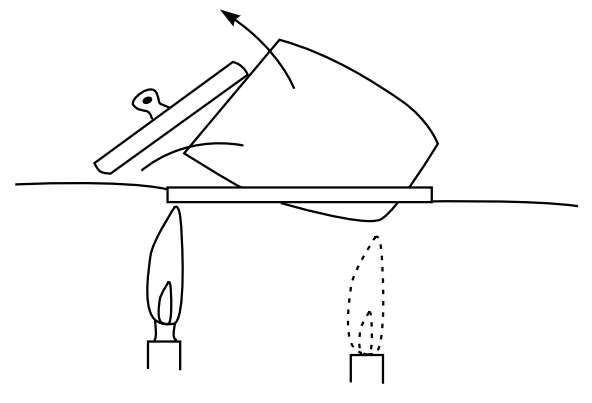
图5-6 沉淀的干燥和灼烧
5.称量 沉淀灼烧完全后,经放至室温,转入干燥器,约30分钟后再称重,直至恒重。灼烧沉淀的过程也可以在高温电热马福炉中完成。此时,一般先将沉淀包的滤纸炭化(加热至黑烟冒尽),再置入高温电热马福炉中灰化。
采用何种灼烧技术,可视实验室的装备决定。若用滤纸过滤,则必须先将滤纸碳化后再加热至无黑色微粒,才将其送入高温炉(也可采用微波炉)灼烧至恒重;而若用玻璃砂芯漏斗进行过漏,则应待沉淀中的溶液抽干,把沾在外壁的水擦干后,再放入电热干燥箱干燥至恒重。
(覃永余)