
下载掌阅APP,畅读海量书库
立即打开



(一)防腐剂用量的有关计算
药物水溶液易被微生物所污染,尤其在含有营养性物质时更是如此。为了保证制剂质量,除在生产制剂的条件和处方设计上考虑排除微生物在制剂中生长的条件外,在制剂中还需加入一定量的防腐剂。
1.不同pH条件下防腐剂的用量
酚类、羟苯酯类和有机酸类防腐剂在溶液中以离子状态存在的部分对微生物细胞并无透膜能力,只有以分子状态存在的部分才可能透过微生物的细胞膜而产生防腐作用。这些防腐剂均为弱酸性物质,它们在水溶液中的解离程度与溶液的pH有关。

式中,[A - ]为防腐剂在溶液中以离子形式存在的“盐”浓度;[HA]为防腐剂在溶液中以游离酸分子形式存在的“分子型浓度”;p K a 为弱酸解离常数的负对数。
因此,一定浓度的防腐剂在不同的pH条件下其防腐效果就不一样。
【例2-19】 假设苯甲酸的防腐有效浓度以游离苯甲酸计算为0.5g·L -1 ,试计算在下列不同pH条件的苯甲酸用量:pH=3.2,pH=5.2,pH=6.2。(已知苯甲酸的p K a =4.2)
解:
当pH=3.2时 lg
 =4.2-3.2=1
=4.2-3.2=1
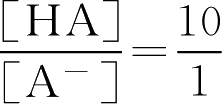
即[HA]10份,[A - ]1份,共11份,其中[HA]应为0.5g·L -1
所以,苯甲酸用量应为0.5×
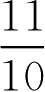 =0.55(g·L
-1
)
=0.55(g·L
-1
)
当pH=5.2时 lg
 =4.2-5.2=-1
=4.2-5.2=-1

所以,苯甲酸用量应为0.5×
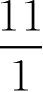 =5.5(g·L
-1
)
=5.5(g·L
-1
)
当pH=6.2时 lg
 =4.2-6.2=-2
=4.2-6.2=-2

所以,苯甲酸用量应为0.5×
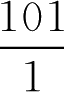 =50.5(g·L
-1
)
=50.5(g·L
-1
)
同理可以计算pH=7.2时,苯甲酸用量应为500.5g·L -1 ,才能起到防腐作用,所以在中性、碱性溶液中不宜使用。
2.表面活性剂存在下微溶性防腐剂的用量
微溶性防腐剂如酚类、羟苯酯类、有机酸类、芳香油类、三氯叔丁醇、苯甲醇等,在微量表面活性剂(如聚山梨酯0.1~0.2g·L -1 )存在下,可稍稍提高其防腐效力,但在表面活性剂用于增溶时,由于防腐剂与表面活性剂之间产生了螯合配位作用,增加了防腐剂的水溶性,降低了脂溶性,从而降低了对微生物细胞膜的通透性,使防腐效力下降,因而应增加防腐剂用量,才能达到防腐作用。
以上情况所用防腐剂用量可按下式计算:
W = A + fB (2-35)
式中, W 为表面活性剂存在下防腐剂的用量,g·L -1 (质量浓度); A 为微量表面活性剂存在下防腐剂的抑菌用量,g·L -1 (质量浓度); B 为表面活性剂质量浓度; f 为配位因素(通过实验求得近似值)。
【例2-20】 表面活性剂聚山梨酯80与防腐剂羟苯甲酯常用于合剂中,羟苯甲酯在微量聚山梨酯存在下的抑菌质量浓度为0.65g·L -1 ,实验求得配位因素为0.058,求聚山梨酯80用量(质量浓度)分别为10g·L -1 、20g·L -1 、30g·L -1 时羟苯甲酯的用量。
解: 将有关数值代入式(2-35)中得
聚山梨酯8010g·L -1 时羟苯甲酯用量为
W 1 =0.65+0.058×10=1.23(g·L -1 )
聚山梨酯8020g·L -1 时羟苯甲酯用量为
W 2 =0.65+0.058×20=1.81(g·L -1 )
聚山梨酯8030g·L -1 时羟苯甲酯用量为
W 3 =0.65+0.058×30=2.39(g·L -1 )
3.已知一种防腐剂的有效浓度,求另一种防腐剂的有效浓度
根据费氏原则,即作用相同的药物,当其溶液饱和度(有效浓度/溶解度)相等时,生理效应也相近,因而当两种防腐剂产生防腐作用的必需浓度 S r 与其溶解度 S 之比相等时,其抑菌效力相近。所以,可以用效能比(有效浓度/溶解度)来比较各种防腐剂的用量。常用防腐剂的相对分子质量、电离度和p K a 值见表2-6。
表2-6 常用防腐剂的相对分子质量、电离度和p K a 值
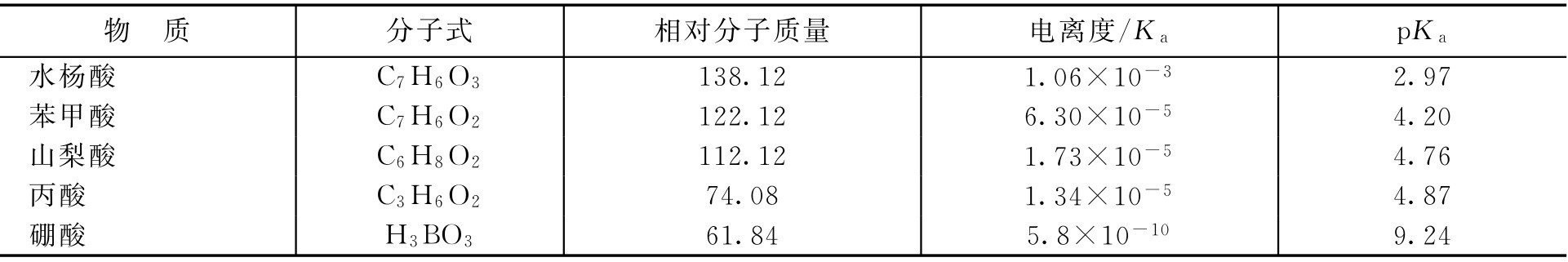

式中, c 为防腐剂有效浓度; S 为防腐剂的溶解度。
当已知某防腐剂的有效浓度 c 时,可根据下式计算另一种防腐剂的有效浓度。

【例2-21】 已知羟苯甲酯的最低有效抑菌浓度为1/1500,求羟苯乙酯和丙酯的有效抑菌浓度。(已知羟苯甲酯、羟苯乙酯、羟苯丙酯的溶解度分别为1/400、1/600、1/2500)

(二)糖浆剂蔗糖投入量的有关计算

糖浆剂是指含药物或芳香性物质的浓蔗糖水溶液。一般要求糖浆剂含蔗糖的质量浓度在450g·L -1 以上。对于西药,蔗糖含量容易控制。而对于中药糖浆剂,相对密度是控制其质量的标准之一,其与蔗糖的投入量、药材的出膏率及成品的体积有关。由于药材的出膏率会受药材的品种、产地、提取条件等因素的影响,所以最终需根据药材浸出浓缩液情况,调节蔗糖的投入量和成品体积,以达到要求的相对密度及蔗糖含量。相对密度是量纲1的量,糖浆的相对密度通常是指糖浆的密度与水的密度(亦称水的体积质量)之比,水的密度是1g·cm -3 或1g·ml -1 ,糖浆的相对密度与糖浆的密度是等值的。
在实际生产过程中很容易测得浓缩液的体积和相对密度,下面介绍如何根据成品要求的相对密度和含蔗糖量来计算蔗糖的投入量和调整最终的体积(即用蒸馏水加至的最终体积)。
根据单糖浆的相对密度1.313(《中国药典》2005年版规定单糖浆相对密度应大于1.30,以下公式仍然适用)和质量浓度850g·L
-1
,计算蔗糖在单位质量单糖浆中所占的体积(ml·g
-1
),用
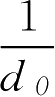 表示,假设有
V
(ml)单糖浆,则
表示,假设有
V
(ml)单糖浆,则
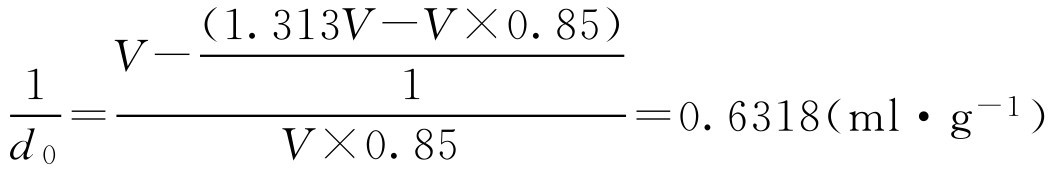
式中, d 0 为单糖浆的密度,g·ml -1 。
上式就是根据体积 V (ml)单糖浆中除去水所占的体积得蔗糖体积,再与蔗糖质量进行比较,即得1g蔗糖在溶液中占的体积为0.6318ml。
为了推导出一般的公式,下面按照糖浆剂制备过程中的两种情况进行讨论。
工艺一:若已知成品体积为 V (ml),密度为 d (g·ml -1 ),蔗糖的含量为 Y (g·ml -1 ),假设制备过程中当提取液浓缩至密度为 d 1 时,只需加蔗糖即得成品,则根据条件求 d 1 :
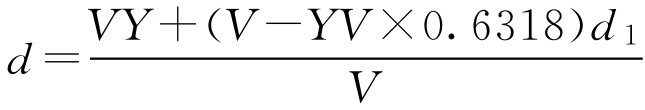
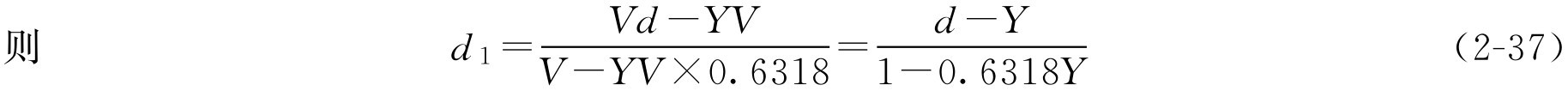
下面求当浓缩液的密度为 d 1 、体积为 V 1 时,只需加入蔗糖量( m )即得体积为 V 、密度为 d 、蔗糖含量为 Y 的成品,则根据此工艺,成品体积等于浓缩液体积 V 1 再加上蔗糖体积,即 V = V 1 +0.6318 YV 整理得
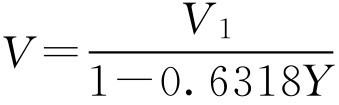
又因为蔗糖的质量等于成品体积与蔗糖含量的乘积即
m = YV
即得蔗糖加入量的公式:

式(2-37)、式(2-38)在实际应用过程中并不方便,现有的工艺设备要将提取液控制到某一相对密度比较难掌握,因此需要建立一个一般性的实际生产中适用的公式。
工艺二:现测得浓缩液密度为 d 2 、体积 V 2 ,工艺要求加入一定量的蔗糖和蒸馏水即得到含糖量为 Y 、密度为 d 的成品,求加入的蔗糖质量( m )和成品最后的体积( V )。
假设 d 1 、 V 1 分别表示用水调整浓缩液后的相对密度及体积(相当于工艺一中 d 1 和 V 1 ),根据质量守恒定律得
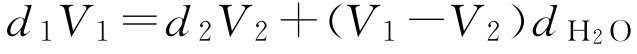
整理得

将式(2-39)代入式(2-38)得
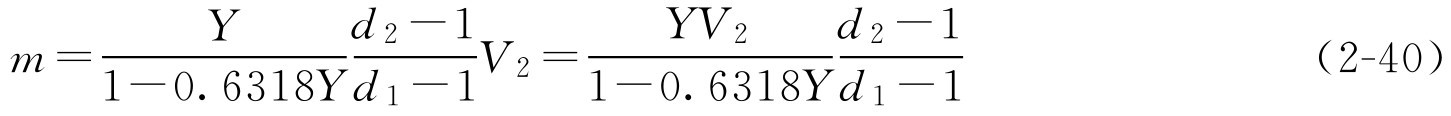
再将式(2-37)代入式(2-40)得
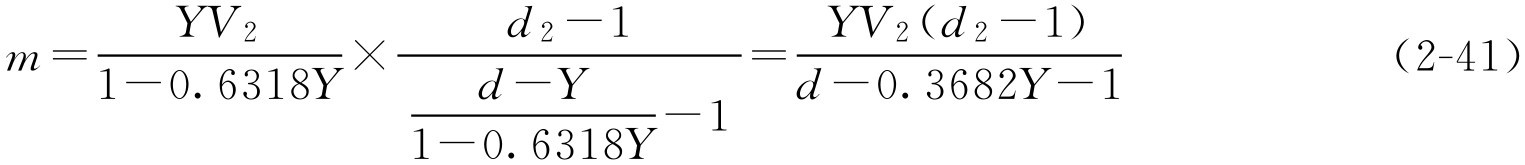
式(2-41)就是通用的蔗糖加入量的公式,在该公式中, Y 、 d 为已知的条件(糖浆剂的一些质量控制内容), V 2 和 d 2 在生产过程中很容易通过密度计及量筒测定得到。
成品的最终体积 V 根据蔗糖的含量求得

即浓缩液中加入计算量的蔗糖溶解后,最后加蒸馏水至体积 V 即可。
【例2-22】 已知某中药糖浆剂成品要求密度为1.29g·ml -1 ,蔗糖含量为0.65g·ml -1 ,现有中间体浓缩液411ml,测得其密度为1.114g·ml -1 ,问需要加蔗糖多少?可得成品多少?
解: 由题意可知: d 2 =1.114, V 2 =411, Y =0.65, d =1.29
按式(2-41)和式(2-42)得:
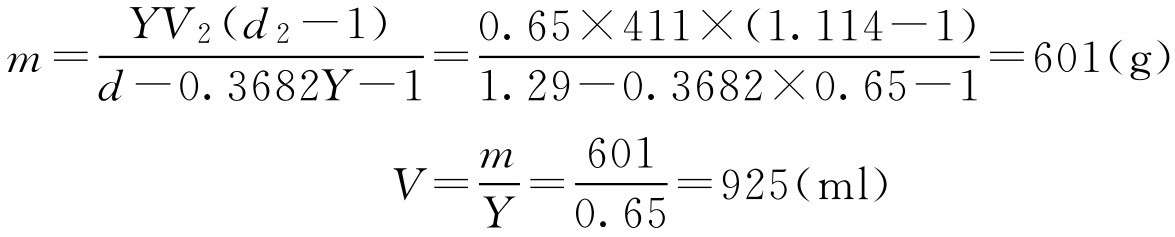
即需要加入蔗糖601g,可得成品中药糖浆剂925ml。
(三)表面活性剂HLB值的有关计算
表面活性物质的分子都具有亲水基和亲油基,为了用定量的方法表示它们亲水和亲油性的强弱,常用亲水亲油平衡值(HLB)来表示。亲水性强的物质具有较高的HLB值,亲水性弱的具有较低的HLB值。根据经验把表面活性剂HLB值的范围定为0~40,其中非离子型表面活性剂HLB值在0~20,将非离子型表面活性剂中完全由亲水基团氧乙烯组成的聚氧乙烯的HLB值定为20,将完全由疏水碳氢链组成的石蜡的HLB值定为0,故HLB值愈低则亲水性愈弱,愈高则亲水性愈强,亲油性则反之。
1.HLB值的计算
(1)天然乳化剂HLB的计算。用已知“所需HLB值”的油类和水加混合乳化剂(系以已知HLB值的合成乳化剂和待测HLB值的天然乳化剂,按不同比例配合)配制成一系列乳剂,经放置后,根据油水分离情况找出最稳定的乳化剂配方。然后按最佳配方中两种乳化剂的配比,求算其中天然乳化剂的HLB值。
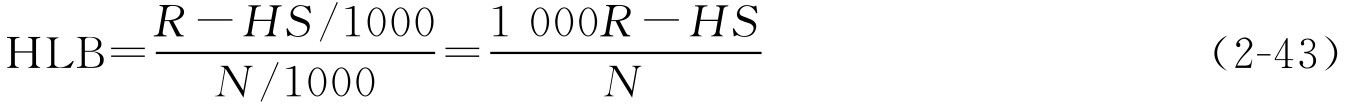
式中,HLB为天然乳化剂的HLB值; R 为油相所需的HLB值; H 为合成乳化剂的已知HLB值; S 为合成乳化剂的质量浓度,g·L -1 ; N 为天然乳化剂的质量浓度,g·L -1 。
【例2-23】 假定天然乳化剂阿拉伯胶的HLB值未知,将阿拉伯胶与合成乳化剂聚山梨酯80分别以675g·L -1 和325g·L -1 的比例制成混合乳化剂,能使水和液状石蜡制得最稳定乳剂,试求阿拉伯胶的HLB值。
解: 聚山梨酯80的HLB值=15.0(H)
液状石蜡所需HLB值=10.5(R)
S =325g·L -1 ; N =675g·L -1 (油/水型)
代入式(2-43)得
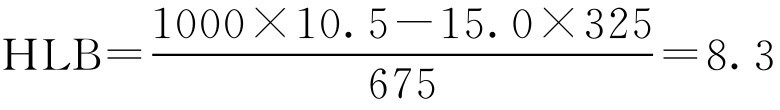
即天然乳化剂阿拉伯胶的HLB值为8.3。
(2)聚氧乙烯型和多元醇型合成表面活性剂的HLB值的求算

式中, M 水 为表面活性剂分子中亲水基团的相对分子质量; M 油 为表面活性剂分子中亲油基团的相对分子质量。
【例2-24】 聚乙二醇分子里没有亲油基团,完全是亲水基团,求其HLB值。
解: 按式(2-44)
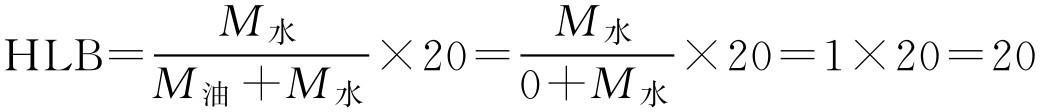
即聚乙二醇的HLB值为20。
(3)大部分多元醇脂肪酸酯如聚山梨酯、脂肪酸山梨坦、单硬脂酸甘油酯等HLB值的求算

式中, S 为酯类皂化价; A 为原料脂肪酸的酸价。
【例2-25】 单硬脂酸甘油酯的皂化价为161,单硬脂酸的酸价是198,求单硬脂酸甘油酯的HLB值是多少?
解: 按式(2-45)
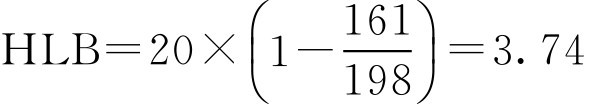
即单硬脂酸甘油酯的HLB值为3.74。
【例2-26】 聚山梨酯20的皂化价为45.5,月桂酸的酸价为276,求聚山梨酯20的HLB值。
解: 按式(2-45)
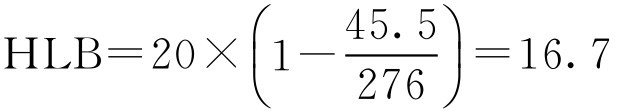
即聚山梨酯20的HLB值为16.7。
2.HLB值的应用
(1)混合乳化剂的HLB值。HLB值具有加成性,无配伍禁忌的混合乳化剂的效用一般比用一种更稳定,将乳化剂A[ m A (g)]和乳化剂B[ m B (g)]混合,混合后的HLB值可按下式计算:
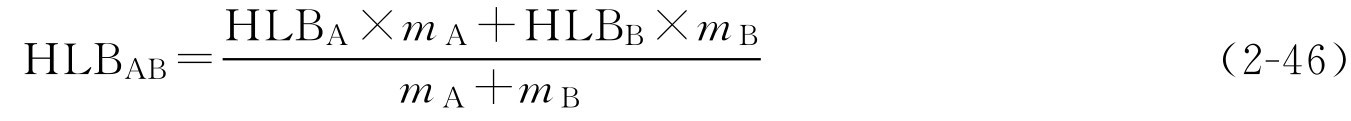
式中,HLB AB 为混合乳化剂的HLB值;HLB A 为乳化剂A的HLB值; m A 为乳化剂A的取量,g;HLB B 为乳化剂B的HLB值; m B 为乳化剂B的取量,g。
对于三种以上混合乳化剂的HLB值可用下式表示:
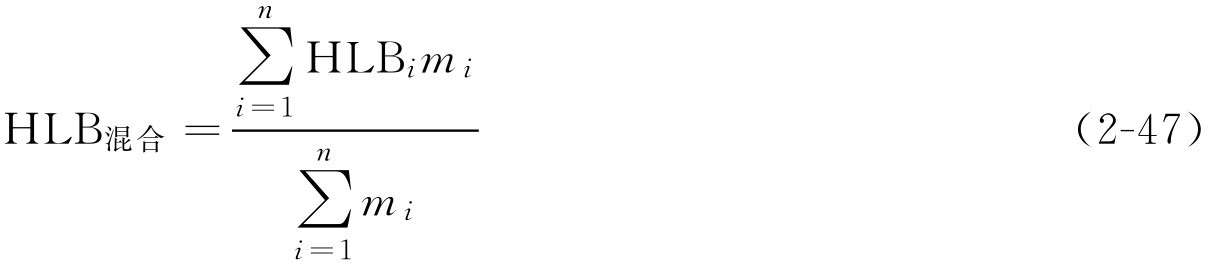
【例2-27】 脂肪酸山梨坦80(HLB为4.3)45g和聚山梨酯80(HLB为15.0)55g混匀后的HLB值为多少?
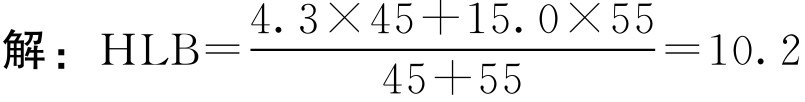
混匀后的HLB值为10.2。
(2)根据油相所需HLB值,求算混合乳化剂各组分用量
如两组分混合乳化剂用量为 a (g),则:

m B = a - m A (2-49)
【例2-28】 试计算下列处方中聚山梨酯80和脂肪酸山梨坦80的用量。

解:
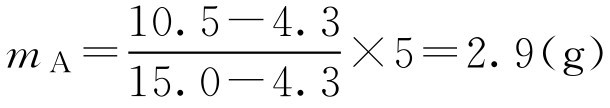
m B =5-2.9=2.1(g)
即应取聚山梨酯802.9g,脂肪酸山梨坦802.1g。
(3)混合油相所需HLB值计算
两种油混合时:
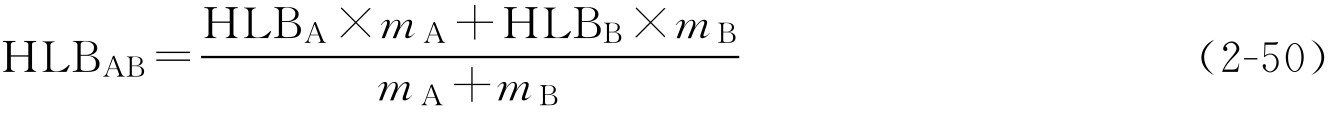
多种油混合时:
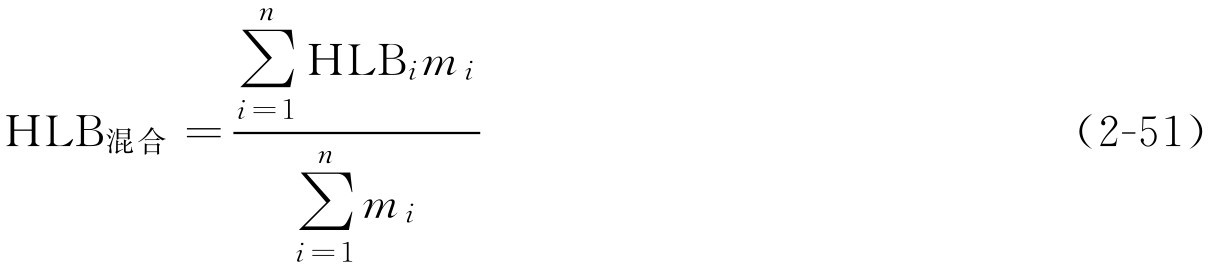
式中,HLB 混合 为混合油相所需HLB值;HLB i 为混合油相中某一种油所需HLB值; m i 为混合油相中某一种油的用量。
【例2-29】 处方
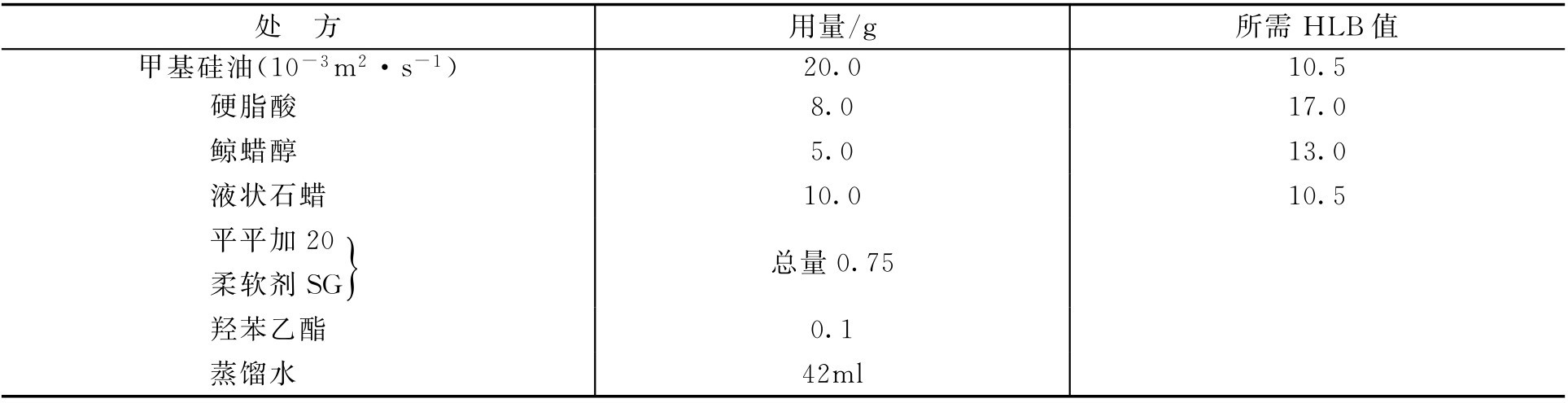
求应该用平平加20和柔软剂SG各多少?(平平加20和柔软剂SG的HLB值分别为16.0和10.0)
解: (1)混合油相所需HLB值为:
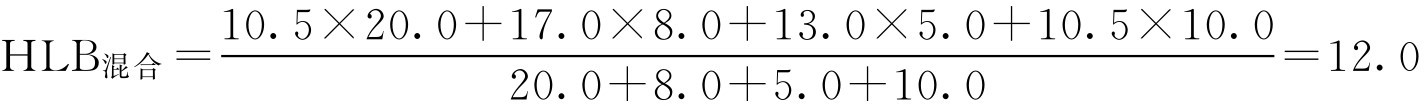
(2)两种乳化剂用量分别为:
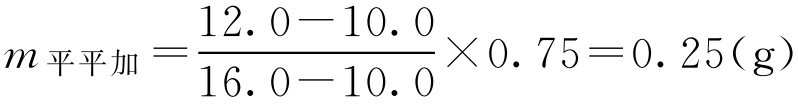
m 柔软剂 =0.75-0.25=0.5(g)
(3)验证。看混合乳化剂的HLB值是否等于混合油相所需HLB值:
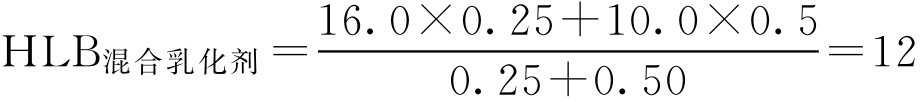
表面活性剂的HLB值范围、乳化剂常用油相的HLB值、常用乳化剂的HLB值分别见表2-7、表2-8和表2-9。
表2-7 表面活性剂的HLB值范围
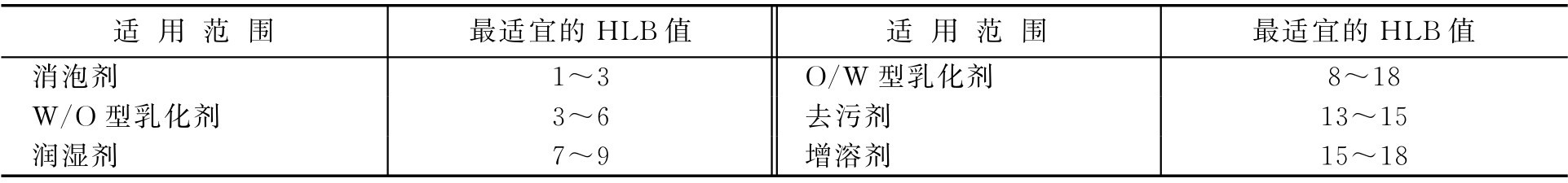
表2-8 乳化剂常用油相的HLB值
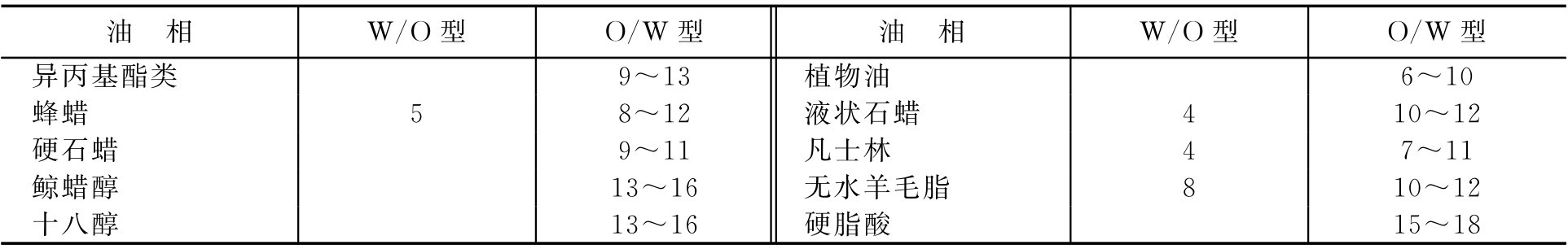
表2-9 常用乳化剂的HLB值
(一)注射剂投料的计算
1.原料不纯时的投料计算

原料理论用量=实际配液数×成品含量(%) (2-53)
【例2-30】 某批维生素B 12 含水质量分数为10%,干燥后维生素B 12 的质量分数为97.0%,欲配100mg·L -1 的注射液10L,应称取多少?
解: 原料理论用量=实际配液数×成品含量(%)=10×100=1000(mg)=1(g)
即:10L需要维生素B 12 1g。
假如成品含量按标示量100%计算:
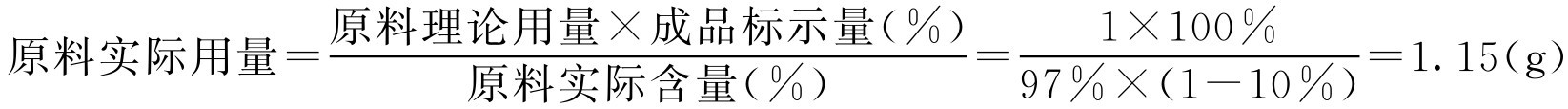
实际配液时,因称量时维生素B 12 极易吸湿,影响成品含量,往往用原包装(1g或2g)直接配制比较方便。
2.根据化学反应式投料
见第一章。
3.含有结晶水药物的投料
见第一章。
(二)等渗溶液的计算
人体内的细胞膜类似半透膜,它允许水分子自由通过细胞膜向高浓一侧移动以达到膜两侧的渗透压平衡。如果注射液渗透压高于或低于注射部位不仅会对注射部位产生刺激性,也会影响其吸收。对于血管内注射,则会对红细胞产生影响,尤其是大容量注射剂(输液),在低渗溶液中,水分子穿过细胞膜进入红细胞内,使红细胞胀破,造成溶血现象。当注入高渗溶液时,细胞内水分渗出而细胞萎缩,但只要注射速度缓慢,由于血液可自行调节使渗透压很快恢复正常,所以影响相对较小。由于以上原因,在设计处方时,对于低渗溶液必须进行调节,常用的渗透压调节方法有以下几种。
1.用Van't Hoff公式计算渗透压
p 渗 = cRT (2-54)
式中, p 渗 为渗透压,kPa; c 为物质的量浓度,mol·L -1 ; R 为摩尔气体常数,等于8.314J·mol -1 ·K -1 ; T 为热力学温度,K。
体液的渗透压为719.2kPa(临床上把709.1~820.5kPa当作正常范围),体温按热力学温度计算为273.2+37=310.2K。
与体液等渗时,任何不电离的药物,其物质的量浓度为:
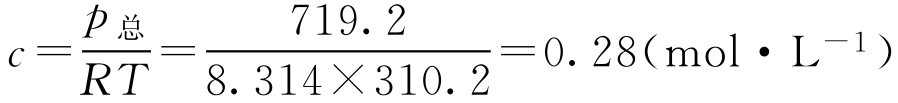
即每升溶液中含0.28mol不电离的药物与体液等渗。
【例2-31】 试计算葡萄糖(C 6 H 12 O 6 )等渗液的浓度。
解: 葡萄糖(C 6 H 12 O 6 ·H 2 O)的相对分子质量为198,其等渗溶液每升应含C 6 H 12 O 6 ·H 2 O198×0.28=55.4(g),即55.4g·L -1 葡萄糖(C 6 H 12 O 6 ·H 2 O)溶液为等渗溶液。
1977~2015年版中国药典的50g/L(即旧称5%)标度葡萄糖注射液,是按含葡萄糖(C 6 H 12 O 6 ·H 2 O)计算的,因此是低渗的。1963年版中国药典50g/L(即旧称5%)标度葡萄糖注射液,在规格项下(4)500ml:25g;(5)1000ml:50g两者后面标注有“(按无水物计算)”,即这两个规格是等渗注射液品种。英国、日本药典各版本的50g/L(即旧称5%)标度葡萄糖注射液均是等渗的,美国药典各版本50g/L(即旧称5%)标度葡萄糖注射液都是低渗的。
2.冰点降低数据法
冰点下降是稀溶液的依数性之一,下降程度取决于溶液中粒子数目,因而两种溶液只要其所含粒子数相等,冰点下降值亦相同,它们渗透压也相同,即等渗。
在药剂工作中,所谓等渗是指药物溶液与血液或泪液的冰点下降值相等而言。
溶液的冰点下降程度Δ T f 与溶液中溶质质量摩尔浓度 m 成正比:
Δ T f = K f m
K f 为一常数,与溶剂有关,如以水为溶剂、非电解质为溶质时 K f =1.86,对于稀溶液,上式可改写成:
Δ T f = K f c (2-55)
式中, c 为物质的量浓度。
如果1L溶液中含有溶质(摩尔质量为 M )质量 m (g),则:

血液和泪液使冰点下降的数值是0.52℃,即:Δ T f =0.52,由式(2-55)、式(2-56)得:

令任一种药物10g·L -1 溶液的冰点下降为 b ,则由式(2-57)得:

溶质为电解质时,因其在水中解离的关系,而需对 K f 值进行校正,应加校正因素 i ,将 i × K f 表示为 L ,则式(2-58)可改写为:

当遇到一种新药,而无现成的10g·L -1 冰点下降值时,可用式(2-59)计算,各种类型化合物的平均 L 值见表2-10。
表2-10 各种类型化合物的平均 L 值

用式(2-57)除以式(2-58)得

当某药的10g·L -1 冰点下降值已知时,配其等渗溶液1000ml所需药量可用式(2-60)计算,如配制等渗溶液 V (ml)可用下式:

式中, b 为药物的10g·L -1 冰点下降值。
如果某药溶液是低渗,需添加其他药物调节等渗时,可用下式:

式中, b 为主药10g·L -1 冰点下降值; ρ 为主药质量浓度; b 0 为调节渗透压添加药物10g·L -1 冰点下降值。
当几种药物配成溶液 V (ml),用另一种药物调节为等渗时,可用下式计算用量:

式中, m 为调节渗透压添加药物的质量; b 0 为调节渗透压添加药物(10g·L -1 )冰点下降值; b i 为处方中各种药物浓度为10g·L -1 的冰点下降值; ρ i 为处方中各种药物的质量浓度。
【例2-32】 欲配制1000ml20g·L -1 盐酸普鲁卡因溶液,需加多少氯化钠,使成等渗溶液?(已知10g·L -1 盐酸普鲁卡因的冰点降低为0.12,氯化钠10g·L -1 浓度冰点下降值为0.578℃)
解:
根据式(2-61)得
m
=
 =43.3(g)
=43.3(g)
即43.3g·L -1 的盐酸普鲁卡因为等渗溶液,20g·L -1 为低渗溶液需补加氯化钠至等渗。
根据式(2-62)得:
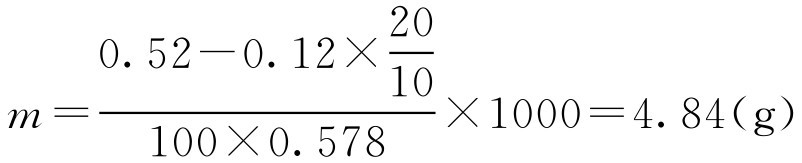
即还需加入4.84g氯化钠才能使1000ml20g·L -1 的盐酸普鲁卡因溶液等渗。
【例2-33】
某一新药为
 A
-
型盐类电解质,相对分子质量为187,试求其10g·L
-1
冰点下降值,如配制20g·L
-1
的溶液100ml,问需添加氯化钠多少以调节成等渗?
A
-
型盐类电解质,相对分子质量为187,试求其10g·L
-1
冰点下降值,如配制20g·L
-1
的溶液100ml,问需添加氯化钠多少以调节成等渗?
解: 该新药的 M r =187,其摩尔质量 M =187g·mol -1 ,按式(2-59)
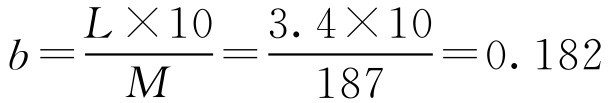
即此药的10g·L -1 冰点下降值为0.182,根据式(2-62)
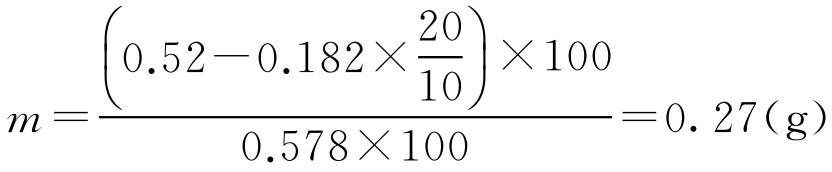
即配制此药20g·L -1 100ml需添加氯化钠0.27g方成等渗溶液。
【例2-34】 硫酸锌眼药水含硫酸锌2g·L -1 、硼酸10g·L -1 ,欲配制500ml,需补加多少氯化钠以制成等渗溶液?
解: 溶液由两种以上药物(除调等渗用药外)配成,可用式(2-63)计算:
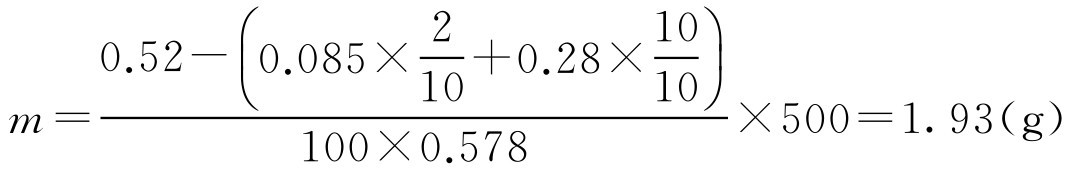
即需补加1.93g氯化钠。
常用药物的10g·L -1 冰点下降值可参阅表2-11。
3.氯化钠等渗当量法
氯化钠等渗当量系指与1g药物呈等渗效应的氯化钠量。可按下式计算配制等渗溶液需添加的药物量:
X =9 V - Em (2-64)
式中,9为生理盐水质量浓度,9g·L -1 ; X 为需加氯化钠的质量,g; V 为欲配制的溶液体积,ml; E 为1g药物的氯化钠等渗当量(可由查表或测定得到); m 为配制用药物的总质量,即为浓度乘以体积。
常用药物不同浓度时冰点下降值见表2-11。
表2-11 常用药物不同浓度时冰点下降值/℃
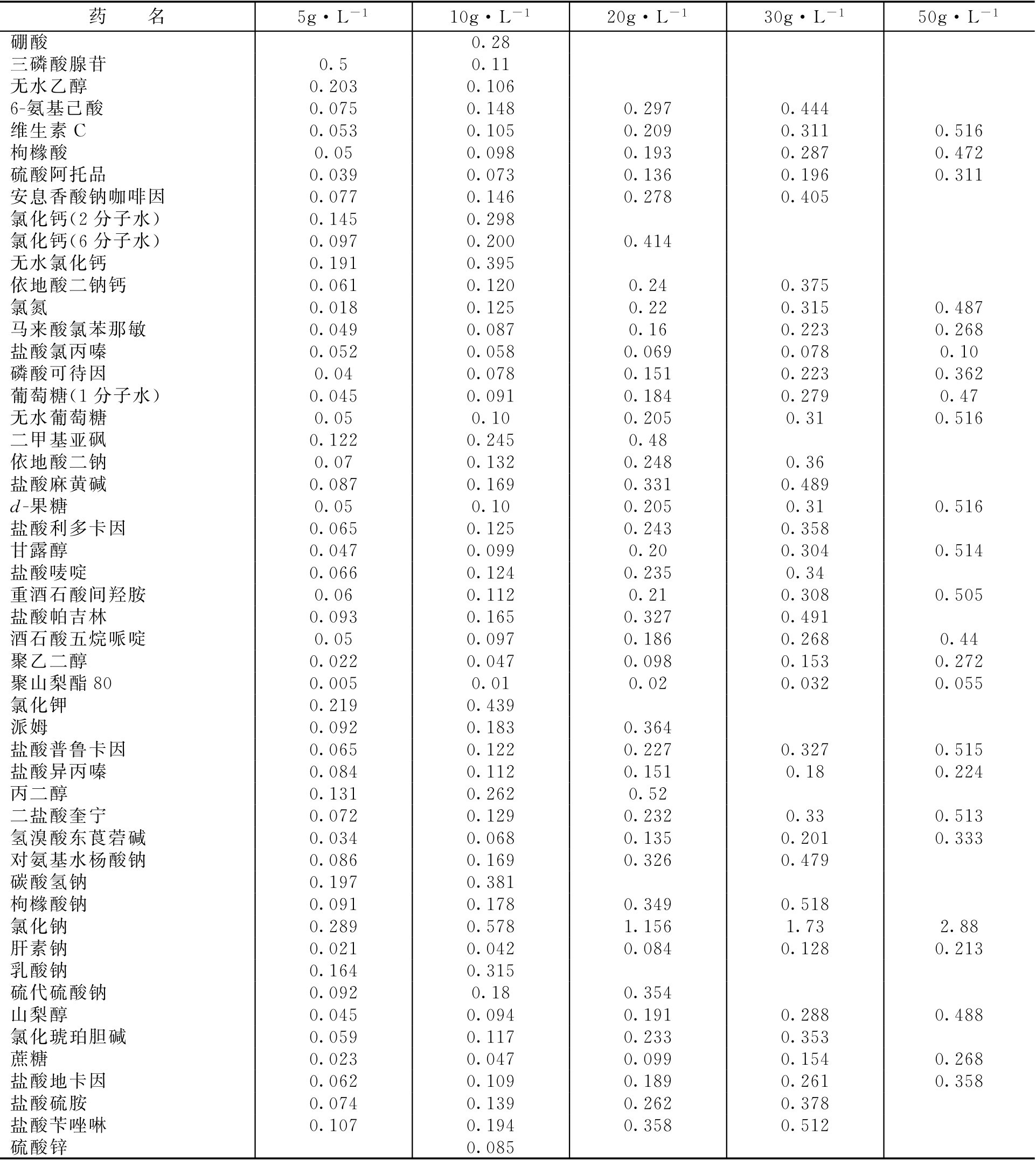
【例2-35】 盐酸普鲁卡因的氯化钠等渗当量为0.21,若配制20g·L -1 的盐酸普鲁卡因0.1L等渗溶液需加氯化钠多少克?
解: 代入式(2-64)得:
X =9 V - Em =9×0.1-0.21×20×0.1=0.48(g)
即需加入0.48g氯化钠才能调节20g·L -1 盐酸普鲁卡因溶液0.1L至等渗。
4.容量值法
容量值,即1g药物配成等渗溶液所需用水的体积,如盐酸麻黄碱的容量值为33.3,即1g盐酸麻黄碱加水到33.3ml即为等渗溶液。
药物的容量值见表2-12。如无容量值,可用氯化钠等渗当量值按下式计算:
表2-12 常用药物的氯化钠等渗当量值和容量值
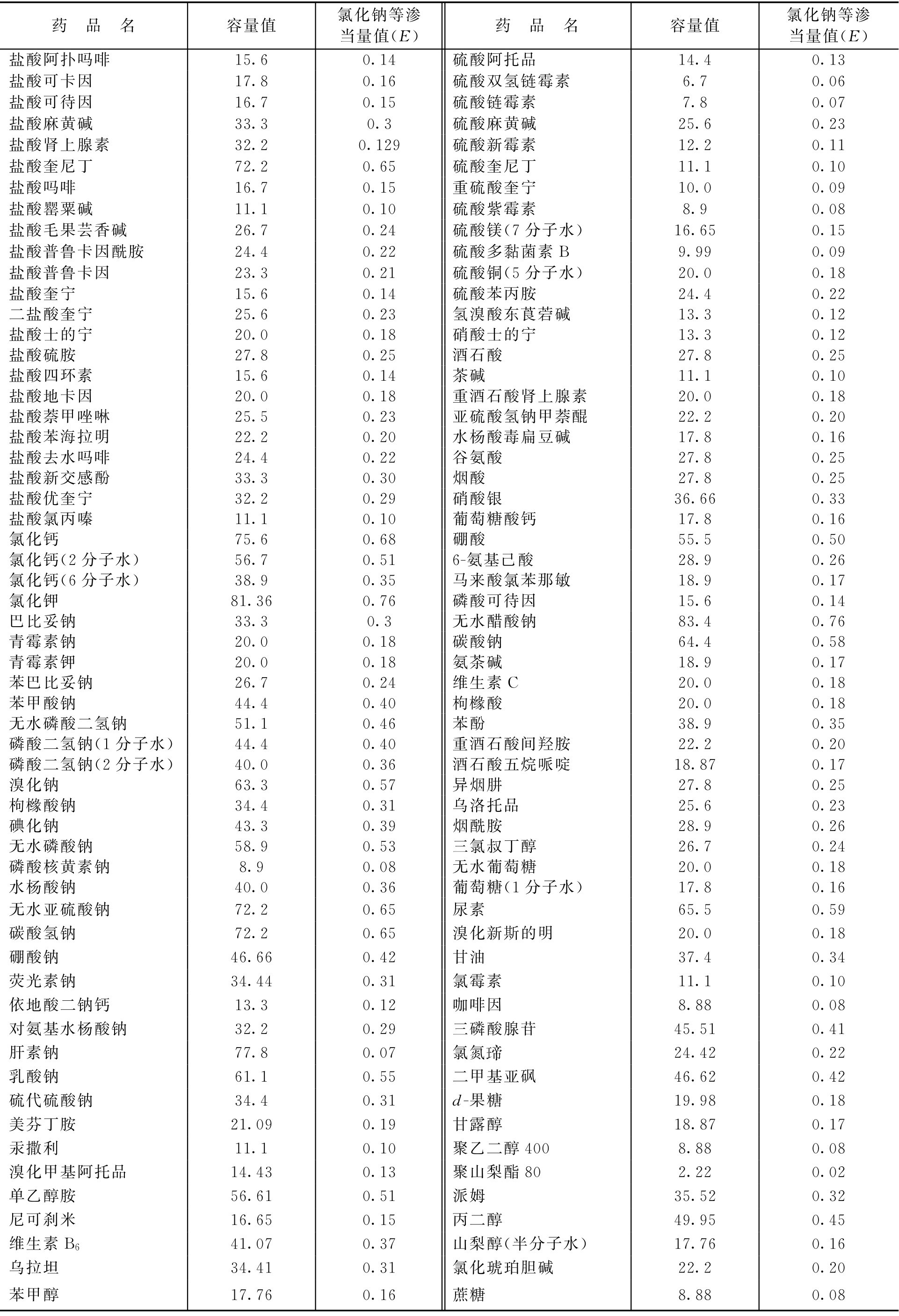
D = E ×111 (2-65)
式中, D 为容量值; E 为氯化钠等渗当量值。
容量值配液法:将药物按容量值配成等渗溶液,再添加适宜的其他等渗溶液(如氯化钠溶液、等渗葡萄糖溶液等)至全量即可。所加其他等渗溶液的量,可用下式计算:
V 加= V (1- ρD ) (2-66)
式中, ρ 为欲配溶液的质量浓度; V 为欲配溶液的体积。
【例2-36】 欲配10g·L -1 盐酸普鲁卡因注射液0.5L,用容量值法配制,需添加生理盐水多少毫升?
解: 查表2-12得盐酸普鲁卡因的容量值为23.3,代入式(2-66),应添加的生理盐水量为:
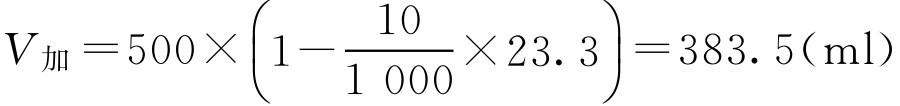
(三)等张度的计算
等渗溶液与等张溶液是两种不同的概念,等渗是物理化学的观点,由溶液的依数性确定,等张是生物学的观点,一般由溶血法测定。如26g·L -1 的甘油溶液与9g·L -1 的氯化钠溶液等渗,但26g·L -1 的甘油100%溶血,所以两者不等张。当然,如果在等渗的情况下不溶血,也是等张溶液。溶血法测定时,将氯化钠溶液从3.6~4.5g·L -1 配成不同浓度梯度,与红细胞共同放置,则会出现不同的溶血现象,一般从4.5g·L -1 开始溶血,3.6g·L -1 左右全溶血。将此系列溶液作为标准比色测定的对照液。另将药物溶液按同样方法与红细胞混合,将两系列溶液分别用比色法测定,如某两种溶液结果相同,即溶血情况相同,则认为它们等张。可用下式计算溶液的等张度或药物的渗透系数。

式中, i 为渗透系数。
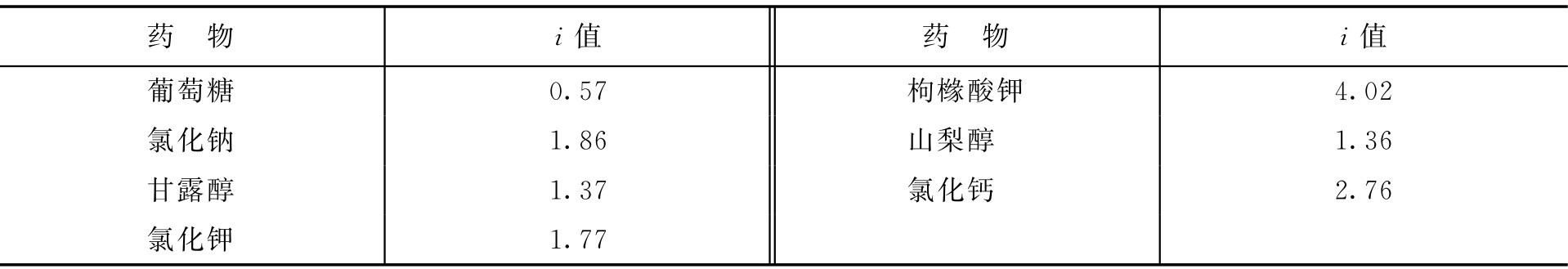
常见药物的渗透系数见表2-13。
表2-13 常见药物的渗透系数
【例2-37】 经溶血法测定得43.6g·L -1 葡萄糖溶液与3.96g·L -1 氯化钠溶液等张,已知氯化钠渗透系数是1.86,氯化钠摩尔质量是58.5g·mol -1 ,葡萄糖(C 6 H 12 O 6 ·H 2 O)摩尔质量是198g·mol -1 ,试根据实验计算葡萄糖的等张质量浓度。
解: 代入式(2-67)计算出葡萄糖与3.96g·L -1 氯化钠相等张的渗透系数,即:

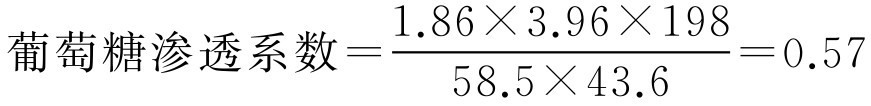
再将葡萄糖渗透系数代入式(2-67)计算出葡萄糖与9g·L -1 氯化钠相等张的质量浓度(即与人体血浆相等张的浓度,简称等张质量浓度),即:

从计算中可看出,物质的渗透系数和摩尔质量在计算过程中都可以通过约分去掉。同时还可看出由
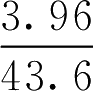 得到的数值实际上是葡萄糖的氯化钠等张当量,由
得到的数值实际上是葡萄糖的氯化钠等张当量,由
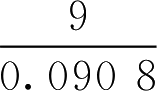 得到的数值是葡萄糖的等张质量浓度。因此溶血法的计算可简化为以下两式:
得到的数值是葡萄糖的等张质量浓度。因此溶血法的计算可简化为以下两式:

所以葡萄糖的等张质量浓度的计算为:

【例2-38】 试计算葡萄糖注射液的等张质量浓度。
解: 葡萄糖(C 6 H 12 O 6 ·H 2 O)的相对分子质量为198, i 为0.57,根据式(2-67)得:
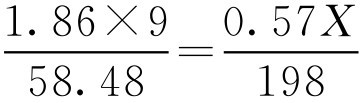
解得 X =99.4g,即葡萄糖(C 6 H 12 O 6 ·H 2 O)的等张质量浓度为99.4g·L -1 。
临床上常用50g·L -1 (等渗)、100g·L -1 (等张)注射液,由以上的计算可知,两者浓度相差近一倍,但由于葡萄糖进入机体后,很快被代谢,不能保持体液张力,所以临床实践常把它们当无张溶液看待。
(四)水醇法制备中草药注射液的含醇量计算
制备中草药注射液常用水提醇沉法来除去水煎液中的淀粉、树脂、蛋白质、黏液质等水溶性物质,加入乙醇量可用下式计算:
C 1 ( V + X )= CX (2-68)
式中, C 1 为水提液需调至的乙醇体积分数,%; C 为加入乙醇的体积分数; V 为水提液(药液)的体积,ml; X 为需加入乙醇的体积,ml。
【例2-39】 用体积分数为95%的乙醇沉淀1000ml麦冬提取液中的杂质,使药液中乙醇体积分数达85%,需加入乙醇多少体积?
解: 根据式(2-68),代入得:
85%×(1000+ X )=95%× X
X =8500ml=8.5L
需加入体积分数95%的乙醇8.5L。
(五)细菌内毒素检查(鲎试剂法)的有关计算
热原主要是微生物产生的一种内毒素。供静脉注射及脊椎腔注射的药物制剂必须进行热原检查。《中国药典》2005年版规定采用家兔法和细菌内毒素检查法检查热原,后者操作简便,它是利用鲎试剂检测供试品中或其表面可能存在的细菌内毒素。常用的是凝胶法和光度测定法。下面将试验中的鲎试剂灵敏度复核、干扰试验涉及的有关计算简介如下。
1.药品细菌内毒素限值( L )的确定
药品细菌内毒素检查是一种限度试验,以确定供试品的细菌内毒素限值。供试品中内毒素含量低于该值,用药后才不致引起致热反应。
药品细菌内毒素限值按下式确定:

式中, L 为供试品细菌内毒素限值标度,以eu·ml -1 、eu·mg -1 、eu·u -1 诸形式表示,eu为内毒素单位; K 为人每千克体重每小时最大可接受的内毒素剂量,以eu·kg -1 ·h -1 表示,注射剂 K =5eu·kg -1 ·h -1 ,放射性药品注射剂 K =2.5eu·kg -1 ·h -1 ,鞘内用注射剂 K =0.2eu·kg -1 ·h -1 ; M 为人用每千克体重每小时最大供试品剂量,以ml·kg -1 ·h -1 、mg·kg -1 ·h -1 或u·kg -1 ·h -1 表示,人均体重按60kg计算,注射时间若不足1h,按1h计算。
对一些没有规定致热原检查剂量的新品种或原料药进行 L 值的计算时, M 值采用人用的最大剂量, L 值按下式确定:
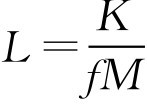
式中, f 为安全系数,一般为3~10。因为致热原检查剂量一般为临床1次用量的3~10倍。
2.鲎试剂灵敏度复核结果的有关计算

式中, λ c 为鲎试剂灵敏度的测定值,eu·ml -1 ; X 为反应终点标度的对数值,lg。
反应终点标度是系列标度递减的内毒素溶液中最后一个呈阳性结果的标度。
当 λ c 在0.5 λ b ~2.0 λ b (包括0.5 λ b 和2.0 λ b , λ b 为鲎试剂的灵敏度标示量)时,方可用于干扰试验和供试品细菌内毒素的检查。
3.供试品干扰试验的有关计算
(1)供试品最大有效稀释倍数MVD

式中, L 为供试品细菌内毒素的限值标度; c 为供试品溶液的标度,当 L 以eu·ml -1 表示时,则 c 等于1.0ml·ml -1 ;当 L 以eu·mg -1 或eu·u -1 表示时, c 的单位需为mg·ml -1 或u·ml -1 ;如供试品为注射用无菌粉末或原料药,则MVD取1,可计算供试品的最小有效稀释标度 c = λ b / L ; λ b 为凝胶法中鲎试剂的灵敏度标示量(eu·ml -1 ),或是光度测定法中所使用的标准曲线上最低的内毒素标度。
(2)干扰试验的结果计算


式中, E s 为用细菌内毒素检查用水配制的内毒素标准溶液反应终点标度的几何平均值; E t 为供试品溶液或稀释液制成的内毒素溶液的反应终点标度的几何平均值。
当 E s 在0.5 λ b ~2.0 λ b 时,且 E t 在0.5 E s ~2.0 E s (包括0.5 E s 和2.0 E s )时,则认为供试品在该浓度下不干扰实验,否则使用更灵敏的鲎试剂,对供试品进行更大倍数的稀释,或做适当处理后重复试验。
【例2-40】 鲎试剂灵敏度复核
解: 设待复核的鲎试剂灵敏度的标示量为0.125eu·ml -1 。
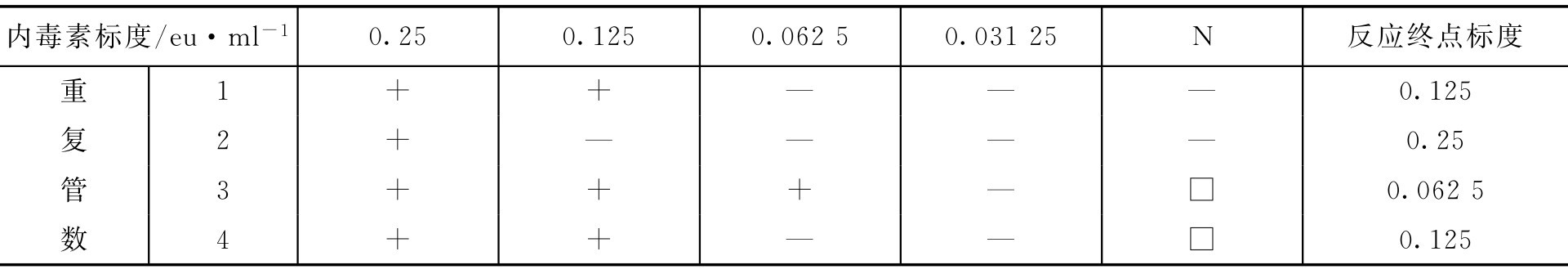
注:N为阴性对照。
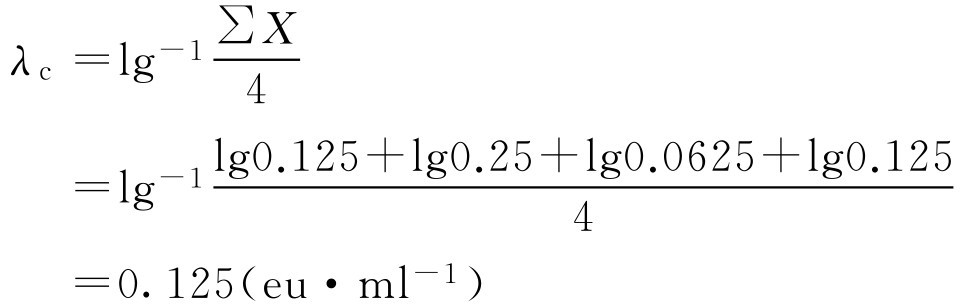
即 λ c 在0.5 λ b ~2.0 λ b 范围内,符合规定。
干扰试验:设所用鲎试剂灵敏度的标示量为0.125eu·ml -1 ,供试品为某注射液(1→4),试验结果如下。
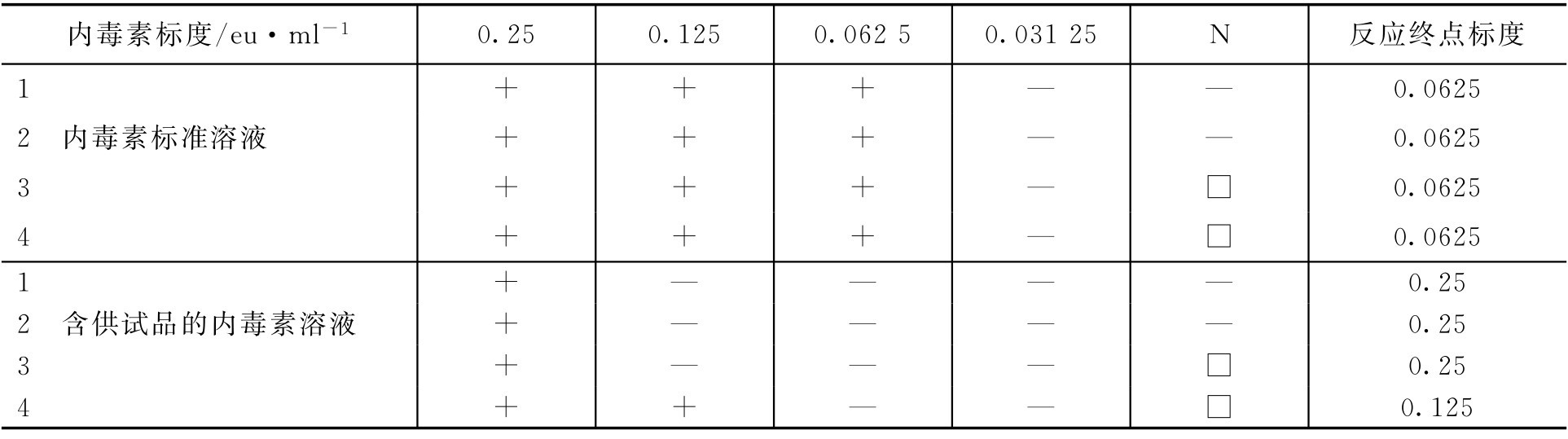
注:N为阴性对照。
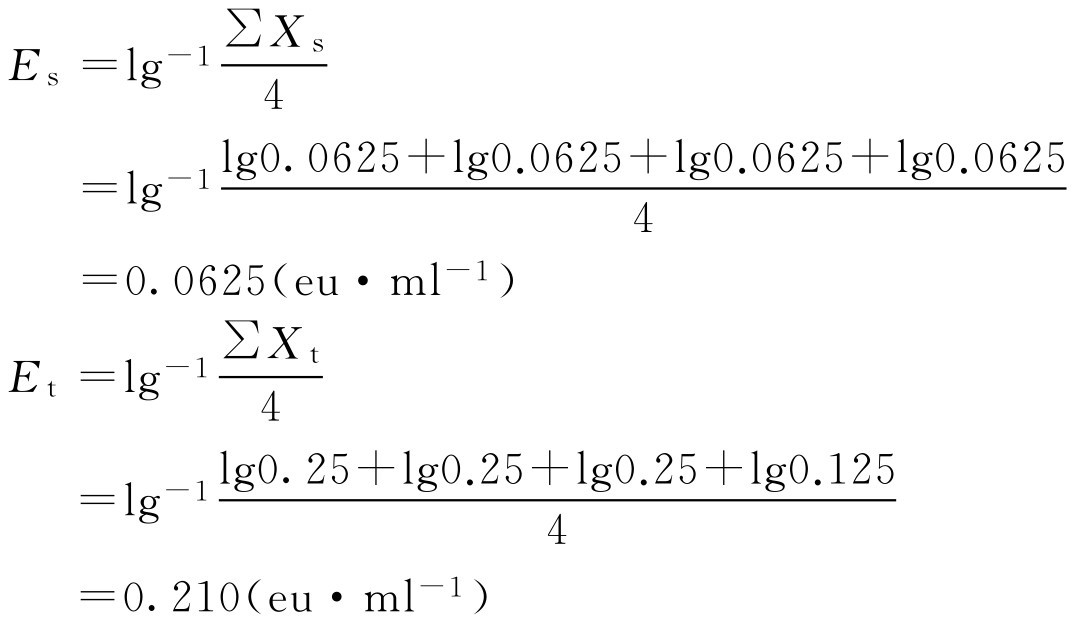
虽然 E s 在0.5 λ b ~2.0 λ b 范围内,但 E t 不在0.5 E s ~2.0 E s 范围,所以该供试品在1→4稀释倍数下有干扰,需进行适当处理后重复试验或对供试品进行更大倍数稀释后进行干扰试验。
(一)粉末药物临界相对湿度的计算
固体药物吸附水分子称为吸湿。如果空气中的水蒸气分压大于药物本身(结晶水或吸附水)所产生的饱和蒸气压,则产生吸湿或潮解。具有水溶性药物的粉末在较低的相对湿度环境时一般不吸湿,但当提高相对湿度到某一定值时,能迅速增加吸湿量,这时的相对湿度称为临界相对湿度(CRH)。水溶性药物都有其固定的CRH值,在CRH值以下时吸湿很少,超过CRH值则显著吸湿,药物的CRH值越大,越不易吸湿,反之则吸湿性大。
散剂通常由几种药物混合制得,水溶性药物的CRH值一般较低,而互相不起作用(如同离子效应、形成复盐等)的水溶性混合物的CRH可用Elder假设计算:
CRH AB ≈CRH A ×CRH B (2-74)
水溶性混合物的CRH值约等于各药物的CRH的乘积,而与各组分的比例无关。
【例2-41】 葡萄糖和维生素C的CRH值分别为82%和71%,若已知两者以2:1的比例混合,试求混合物的CRH。
解: 根据式(2-74)得CRH 混 =82%×71%=58.2%
计算得混合物的CRH值为58.2%,实际测得值为57%,基本相符。
(二)中药丸剂起模用粉量的计算
泛制法制备丸剂,起模是泛丸成型的条件,起模用的粉量除考虑处方中各成分的性质外,还应注意到丸剂的大小和投料的数量,根据生产实践经验,起模用粉量可按下式计算:

式中, X 为起模用粉量,kg; D 为药料总量,kg; C 为成品水丸100粒干重,g。
【例2-42】 现有500kg香砂养胃丸粉料,要求制成每100粒重为10g的水丸,需取多少粉料起模?
解:
X
=
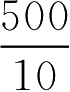 =50(kg)
=50(kg)
即需要50kg粉料起模。
以上为经验式,一般吸水量大的、质地松脆的粉末,起模用粉量应增加,而吸水量少、质地黏韧的粉末则相反。
(一)片重及其计算
片剂的片重与标示量不同,标示量是指主药的含量,片重是指主药和辅料之和,片重准确才能使主药含量准确,在制片过程中,从不同的角度测算,片重有以下几种表示方法。
理论片重:处方中主药加辅料之和。
计算片重:在颗粒制成后,按颗粒中主药含量计算。
实际片重:压片后,成品片重。
平均片重:按药典方法,将若干片的实际总重平均而求得的片重。
在实际生产中主要对“计算片重”进行计算,对于化学药品片剂或已经提得有效成分并能进行含量测定的中草药片剂,其“计算片重”可按下列方法计算。
1.按颗粒中主药含量计算片重

【例2-43】 已知乙酰螺旋霉素片每片含乙酰螺旋霉素0.1g,制成颗粒后,测得颗粒中的含药质量分数为48.5%,本品含乙酰螺旋霉素应为标示量的95.0%~105.0%,试计算片重范围。
乙酰螺旋霉素处方: 每10万片批用量
乙酰螺旋霉素 10.0kg
淀粉 10.8kg
干淀粉(外加) 60g
乙醇40%(体积分数) 适量
硬脂酸镁 160g
解: 已知每片含主药量0.1g,颗粒中含主药质量分数为48.5%,主药允许误差范围为95.0%~105.0%。压片前需加入的辅料为干淀粉和硬脂酸镁,则:
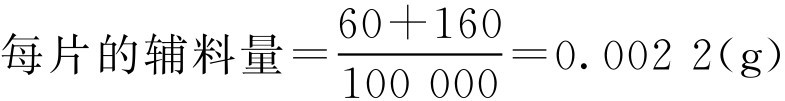
片重根据式(2-76)计算得:
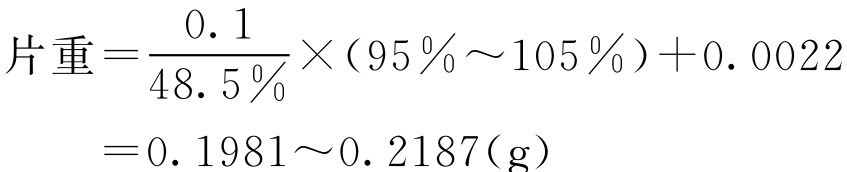
即乙酰螺旋霉素片的片重范围为0.1981~0.2187g。
2.按颗粒质量计算片重

该公式适用于原料损耗量已计入制粒过程中的情况。
【例2-44】 已制得对乙酰氨基酚缓释干燥颗粒94.5kg,压片前加入硬脂酸镁3.0kg,混合压片,共压160000片,试求片重。
解: 根据题意代入式(2-77)得:
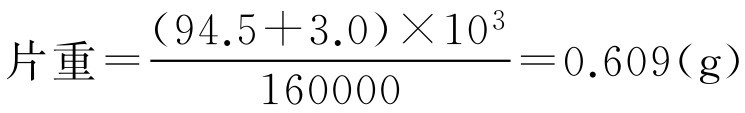
即每片片重为0.609g。
以上两公式适用于原料药,对于中药材,除少数采用有效成分外,多采用浸膏或半浸膏加辅料制粒、干燥、压片制得。对于前者,片重的计算用式(2-76)、式(2-77);对于后者,根据具体情况采用下列相应的公式。
3.按单服颗粒质量计算片重
如果药料的片数及片重未定时,可先称出颗粒总质量相当于若干单服质量,再根据单服质量的颗粒重来决定每服的片数,求得每片的质量。

【例2-45】 有可供服用200次的中草药10kg,经煎煮浓缩后制成干颗粒480g,加入润滑剂、崩解剂共20g后压片,要求每次服5片,计算片重应为多少?
解: 根据式(2-78):
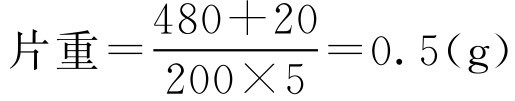
即此中药片剂的片重为0.5g。
4.按原药材服用量及药材提取后所得浸膏量计算片重

【例2-46】 某中药每日服用量为45g,现有20kg,经煎煮后制成浸膏质量为400g,如果按照每日服用6片(每日3次,每次2片)计算,求该中药的片重。

5.浸膏+原粉制得片剂的片重计算
很多中草药片剂在生产过程中常将部分药材提取浓缩成浸膏,而将另一部分药材磨成细粉,将两者混合再加适宜辅料压制成片。

【例2-47】 欲制备每片含原药材1g的穿心莲片1万片,所用原辅料分别为:穿心莲10kg,硬脂酸镁0.03kg。制备时将3kg穿心莲磨成细粉并过80目筛,剩余7kg用水煎煮浓缩成浸膏,若水煎液出膏率为15%,膏中含总固体质量分数为70%。将原粉与浸膏混匀后于60℃干燥,最后加入硬脂酸镁压片,计算穿心莲片的片重。
解: 根据题意,参照式(2-80)代入得:
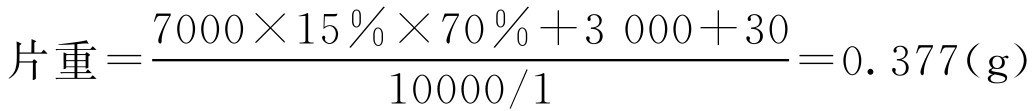
即该穿心莲片的片重为0.377g。
6.中限范围的计算
片剂生产中常用“中限”来控制片重的计算范围值。生产过程中若含量高于或低于中限时则必须调整片重。中限的计算公式如下:


【例2-48】 某药片主药含量为0.2g,含量允许误差范围为±10%,试计算中限范围值。
解: 主药含量低限=0.2×90%=0.18(g)
主药含量高限=0.2×110%=0.22(g)
中限低限=0.18+
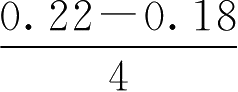 =0.19(g)
=0.19(g)
中限高限=0.22-
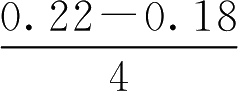 =0.21(g)
=0.21(g)
中限范围为0.19~0.21g,即生产过程中应调节片重将主药的含量控制在该范围内,以确保制得的片剂主药含量符合质量标准。
(二)固体制剂包衣的有关计算
包衣是在制剂成型(片剂、胶囊、颗粒、小丸等)表面再包裹上适宜材料的衣层。随着对包衣认识的深入和高分子材料的发展,从最早掩盖不良臭味的糖衣到目前广泛使用的用于提高药物稳定性的薄膜衣和肠溶衣,选用不同的包衣材料还可达到定位和缓释目的,所以现在包衣技术应用得越来越广泛。在薄膜包衣和肠溶包衣中必须达到一定厚度才能起到防湿或肠溶等目的。衣料的用量与底层的表面积有关,常以每平方厘米表面积制剂聚合物干重(mg)来表示衣料的用量。假设将片剂、胶囊看成圆柱体,以下剂型的表面积可通过下列简化式计算。
片剂: S=π(dh+0.5d 2 ) (2-83)
椭圆形胶囊、片剂: S=πdh (2-84)
球形 (微型片、微丸、颗粒): S=πd 2 (2-85)
式中, S 为单个制剂的表面积,mm 2 ; d 为直径,mm; h 为制剂高度,mm。
上面是面积的简化计算,可用下式精确计算片剂的表面积:
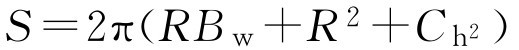
式中, R 为半径; B w 为片剂厚度; C h 为片剂凸出部分高度。
由于片高
H
=
B
w
+
C
h
,而片厚度和片凸出部分高度难以测量,可以用凸半径(
C
r
)计算片剂凸出部分高度
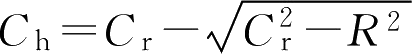 。所以得到:
。所以得到:
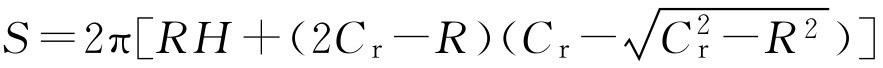
计算制剂的表面积后,用下式计算包衣材料的用量,常用包衣增重百分数表示:

包衣聚合物用量(g)=包衣增重×欲包衣制剂的总质量 (2-87)
式中, S 为待包衣制剂的表面积; L 为每平方厘米表面积所需的薄膜衣干材料用量,mg; m 为单个制剂的质量。
一般薄膜衣层厚度约为8μm,即可掩盖药物的不适臭味,相当于每平方厘米的表面需要1mg的干衣料。包隔离层薄膜衣厚度只需2~5 μ m,相当于每平方厘米的表面需要0.2~0.6mg的干衣料。若为增加制剂储存期间的稳定性,起防护目的,则每平方厘米的表面需要1~2mg的干衣料。肠溶衣每平方厘米的表面需要3~5mg的干衣料。对于一些起特殊定位作用的衣层,可通过实验确定适宜的用量范围。
【例2-49】 某凸形片直径为7mm,厚度3.6mm,片重140mg,用肠溶材料Eudragit L100包肠溶衣,已知聚合物的用量 L 为4mg·cm -2 ,试计算2.5kg素片需用的聚合物的量。
解:根据已知条件先求素片的表面积 S ,代入式(2-83)
S =π( dh +0.5 d 2 )=3.14×(7×3.6+0.5×7 2 )=156(mm 2 )=1.56(cm 2 )
再代入式(2-86)、式(2-87)
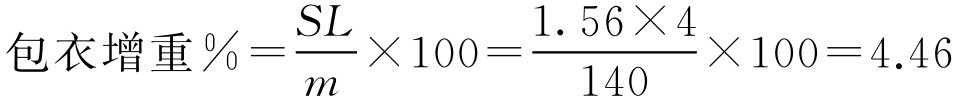
所以聚合物的用量=4.46%×2500=112(g)
即需要112g聚合物Eudragit L100来包肠溶衣。
(三)片剂溶出度测定的计算
评价固体剂型如片剂、胶囊剂等吸收程度的最有效办法是测定人体内的生物利用度,但生产上不可能每批都这样做。而对固体制剂而言,溶出是影响吸收的重要因素,即固体制剂的溶出速率在一定程度上能反映药物的吸收情况,因此往往用体外溶出度(对于缓控释制剂称释放度)作为考察固体制剂内在质量的指标。
片剂溶出度是指药片在规定的介质中(人工胃液或人工肠液),主药成分的溶出速率和程度。一些难溶性药物若不易从制剂中释放出来或药物的溶出速率极缓慢,则药物的生物利用度可能很差;另外,某些药理作用强、治疗指数小的药物,如溶出速率过快,吸收迅速容易产生明显的不良反应或毒性,因此这些药物都需要将其制剂的溶出度控制在一合适的范围内。2005年版药典二部附录ⅩC规定采用转篮法(第一法)和桨法(第二法和第三法),在规定时间取样后按照药品项下规定的方法测定药物浓度,再按照下式计算药物溶出百分率或累积药物溶出百分率。
1.溶出百分率的计算

式中, c 为在规定时间取样后按规定的方法测定的药物浓度。
累积药物溶出百分率的计算也可用下面的修正式:
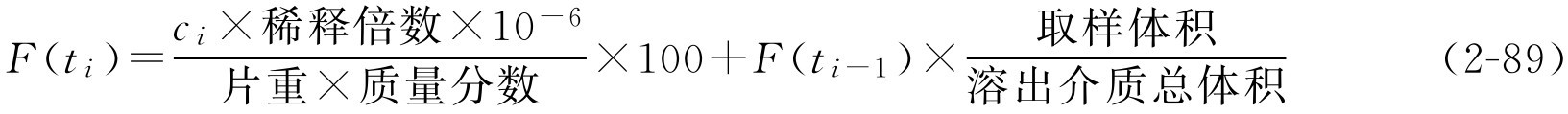
式中, c i 为第 i 次取样时间的药物浓度; F ( t i )表示第 i 次取样时间内药物的累积溶出百分数; F ( t i- 1 )表示 i 前一时刻取样间隔内的累积溶出百分数。
式(2-89)也是一个简化公式,如果要精确计算累积溶出百分率,可用下式:

式中, c n 为 n 次取样药物浓度。
2.溶出参数的计算
测定固体剂型一系列时间药物的溶出百分率后,对实验数据进行处理,获得若干参数用以描述药物或药物制剂在体外溶出的规律,或作为规定制剂的控制指标等。寻求特性参数的方法主要有以下几种。
(1)单指数模型。药物溶出百分率与时间关系符合单指数方程:
y = y ∞ (1-e - kt ) (2-91)
式中, y 为 t 时间的累积溶出百分率; y ∞ 为药物溶出的最大量,通常为100%或接近100%。
将式(2-91)整理取对数得:

用lg( y ∞ - y )对 t 作图为一直线,用一元线性回归求直线回归方程,从直线斜率求出 k , k 值大小可反映溶出速率的快慢。将 y =50%代入回归方程,可求得释放50%所需的时间 t 0.5 。
【例2-50】 已知有两种不同厂家生产的含同样药物的缓释片剂经释放度研究累积释放百分率结果见表2-14,试比较两种片剂释放药物的快慢。
表2-14 不同时间的药物累积释放百分率
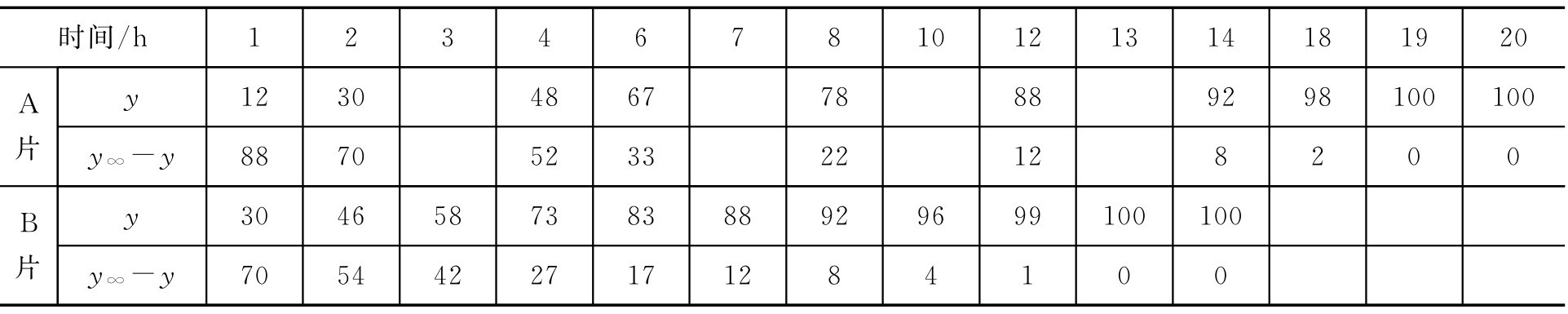
解: 根据式(2-92),以lg( y ∞ - y )对时间 t 进行线性回归,A片剂与B片剂的方程分别为
A片: y =-0.0902 x +2.0641 解得: k A =-2.303×(-0.0902)=0.208
B片: y =-0.1572 x +2.0866 解得: k B =-2.303×(-0.1572)=0.362
由上可知 k B > k A ,即B片释放速率大于A片。
(2)对数正态分布模型。药物释放速率以单指数模型拟合时,在半对数坐标纸上各点若不成直线,可以试用对数正态分布模型:
Y = φ [(lg t - μ )/ σ ] (2-93)
式中, σ 、 μ 为对数正态分布模型的参数, μ 为对数均数, σ 为对数标准差,若制剂的溶出百分率符合对数正态分布模型,则 σ 、 μ 可以反映溶出过程的特征。通常 σ 、 μ 值大,溶出速率缓慢。亦可用均数 m 和标准差 s 表示, m 值大的制剂药物溶出缓慢。
累积溶出百分率用对数正态分布模型拟合的方法如下。
先求出各时间( t )的累积溶出百分率 Y ,再在对数正态分布概率纸中以正态分布坐标(即纵坐标)为累积溶出百分率、对数坐标(即横坐标)表示时间作图。如果各点能连成直线,即表示该制剂的释放规律符合对数正态分布模型。确定各点在对数正态分布概率纸上成一直线,即可求出 σ 、 μ 、 m 和 s 各参数。在图上查出直线在纵坐标上0.5和0.16(或0.84)的对应横坐标值,用 t 0.50 、 t 0.16 或 t 0.84 表示,计算 σ 、 μ 及 m 、 s 的公式如下:
μ =lg t 0.50 (2-94)
σ =lg t 0.50 -lg t 0.16 (2-95)
m =lg -1 ( μ +1.151 σ 2 ) (2-96)

【例2-51】 某药厂生产的阿司匹林片用转篮法测得该片的溶出速率数据见表2-15。求 μ 、 σ 、 m 、 s 。
表2-15 阿司匹林片的溶出速率数据
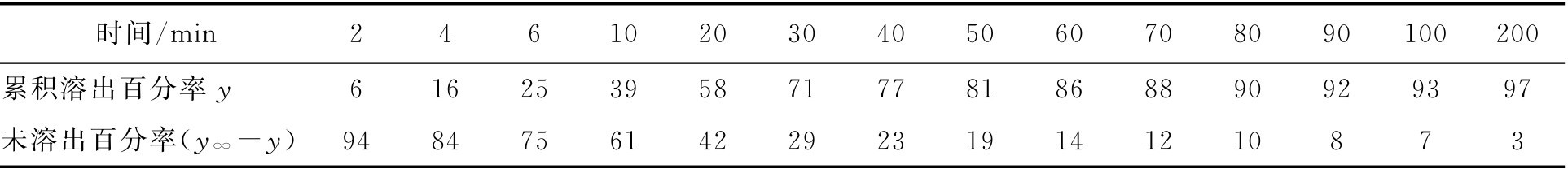
解: 设 y ∞ 为100%,则未溶出百分率为 y ∞ - y ,以 y ∞ - y 值对 t 在半对数坐标纸上作图得一条曲线。该片的释放规律不符合单指数模型。将上述数据在对数正态概率纸上以 Y-t 值作图,连接各点为一直线,见图2-4所示。图中纵坐标为50%、16%处所对应的横坐标为15.2和4.1,即 t 0.50 =15.2min、 t 0.16 =4.1min,将其代入式(2-94)、式(2-95)计算得 μ =1.1818、 σ =0.5691,再将 μ 、 σ 代入式(2-96)得 m =35.85,再将 m 代入式(2-97)得 s =76.63。
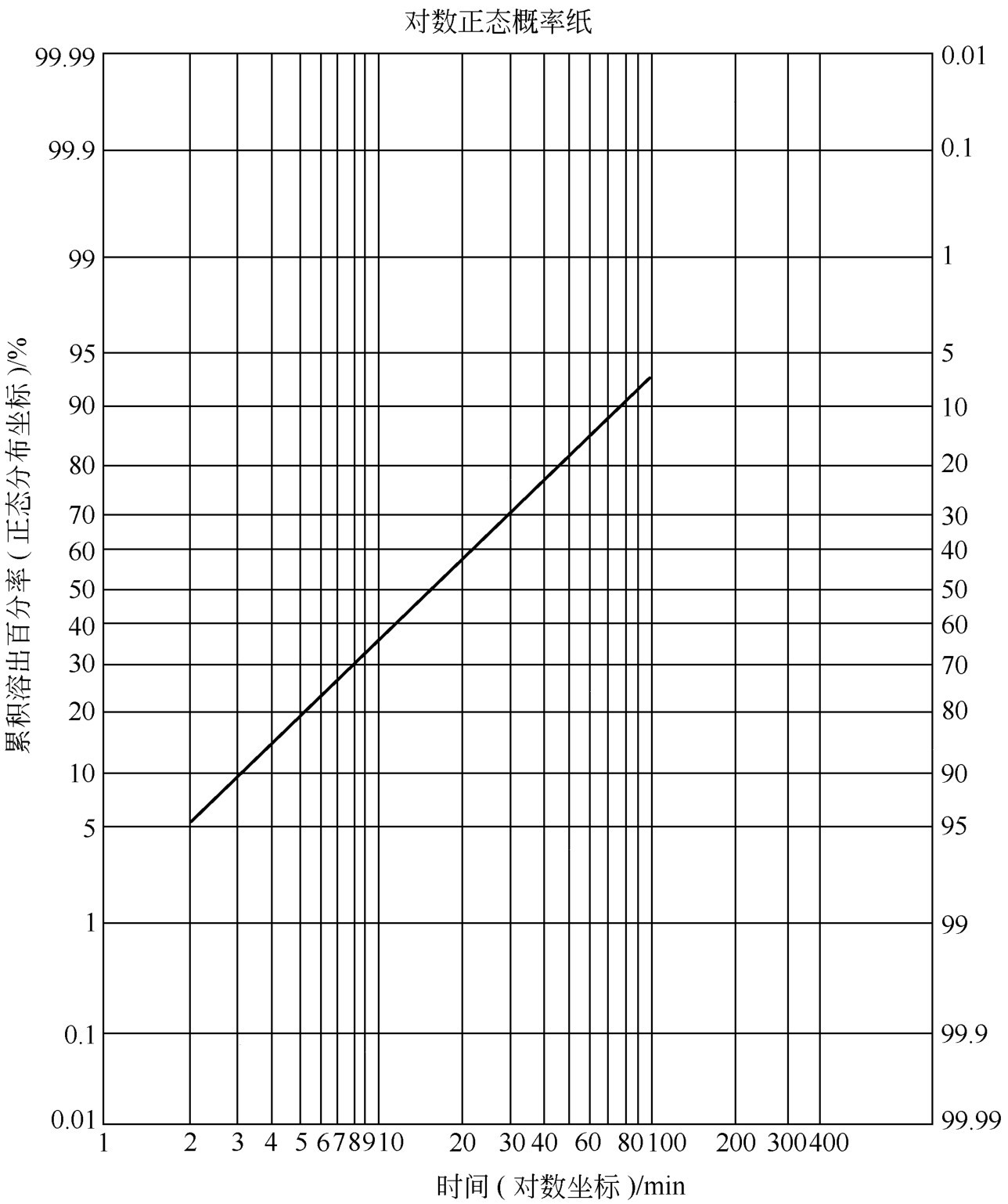
图2-4 阿司匹林累积溶出百分率在对数正态分布纸上作图
(3)威布尔(Weibull)分布模型。见第五章第二节中的Weibull分布。
3.溶出曲线比较
上述有关溶出度评价方法通过制剂溶出度数据分别求算特征性参数(如 k 、 t 0.5 等)后进行差异比较,难以较全面反映溶出全过程的差别。目前国外已经广泛采用溶出曲线比较的方法(又称数学比较法)进行指导处方设计、工艺参数变更、溶出度评价等。由于直观上很难判断溶出曲线之间的不同,因此国际上采用两个溶出系数:变异因子( f 1 )与相似因子( f 2 )来定量评价参比制剂与试验制剂溶出曲线之间的差别。评价两制剂相似性的标准是:当 f 1 在0~15之间,或 f 2 在50~100之间,且在任何时间点溶出度的平均误差均不超过15%,则表明两种制剂的溶出度相似或相同。美国食品药品监督管理局(FDA)在1999年的《口服固体制剂生物利用度和生物等效性研究指南》中推荐使用相似因子法评价其溶出度,以确定仿制药品与对照药品的溶出度差异。
f 1 和 f 2 值的计算公式如下:
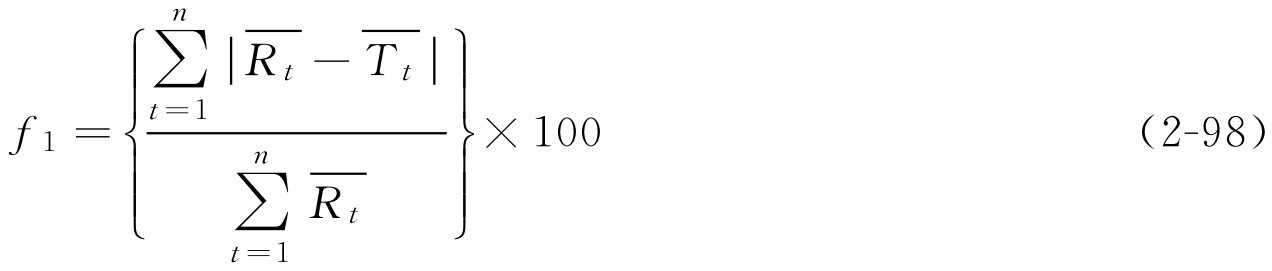
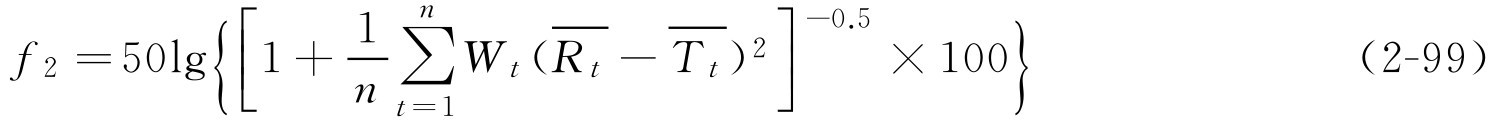
式中,
n
为取样点数目;
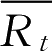 和
和
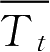 分别为在
t
时间点的参比制剂与试验制剂平均累积溶出百分率;
W
t
为不同时间点的权重系数,可根据不同时间点溶出度的重要性设定不同的值,当无法确定大小时,
W
t
可设为1。
分别为在
t
时间点的参比制剂与试验制剂平均累积溶出百分率;
W
t
为不同时间点的权重系数,可根据不同时间点溶出度的重要性设定不同的值,当无法确定大小时,
W
t
可设为1。
【例2-52】 如某单位研制的双嘧达莫缓释片与进口制剂的不同时间累积溶出百分率如下:

试用相似因子( f 2 )评价两制剂是否相似。
解: 根据式(2-99)
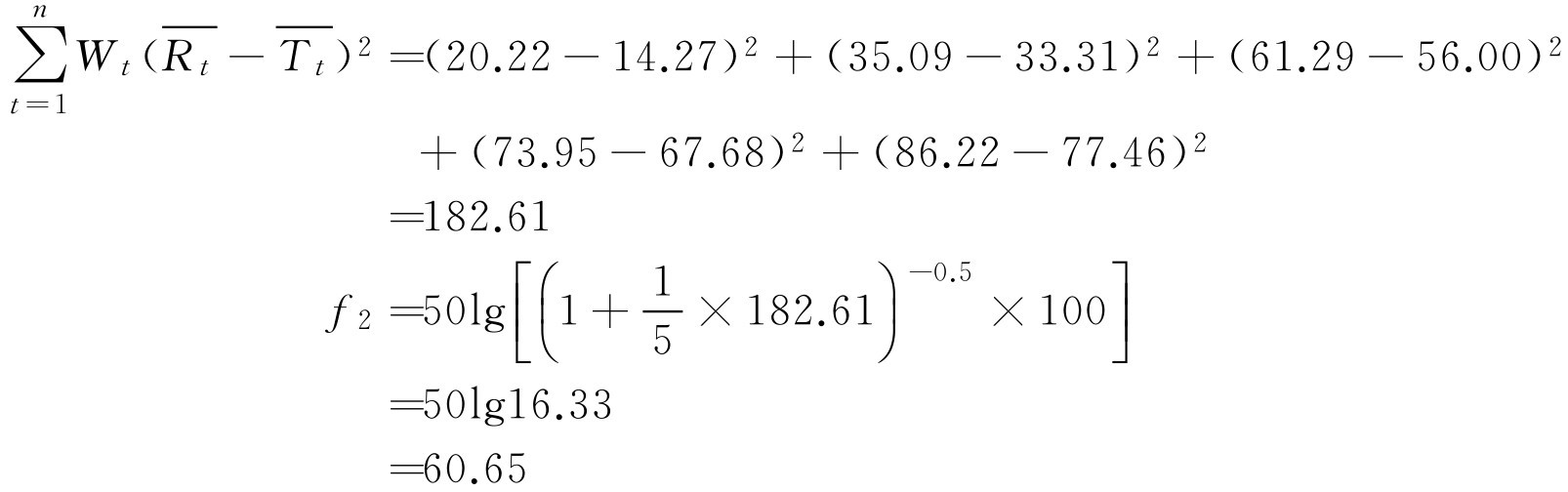
由计算结果可知 f 2 为60.65,在50~100之间,表明研制的双嘧达莫缓释片与进口制剂的释放度相似。
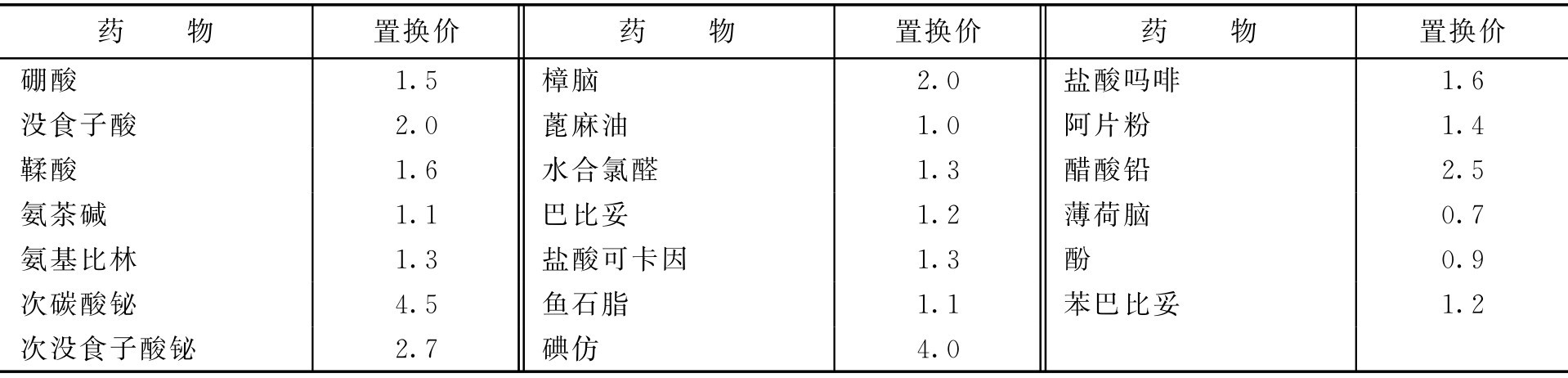
检剂置换价为主药质量与同体积基质质量之比,即

测置换价的方法主要有以下两种。
1.经典法(质量法)
在制备栓剂时,用同一模型所制栓剂的体积虽相同,但其质量则随基质与药物的密度不同而不同,通常均以基质可可豆脂为标准。如主药的密度与可可豆脂的密度相同时,药物可置换等重的可可豆脂;但如主药的密度较可可豆脂大2倍时,则主药仅占等重可可豆脂容量的一半。主药的质量与同体积可可豆脂质量的比值称为置换价。则置换价的求算或已知置换价求基质的用量可按下式计算:

式中, E 为置换价; W 为每颗栓剂中的主药质量; G 为纯基质的质量(一颗栓剂中完全是基质,没有主药); M 为每颗栓剂的质量。
【例2-53】 栓模标量为3g(按可可豆脂计),将药物1.5g与可可豆脂相混合,倒入栓模中,测得栓剂的质量为3.7g,此药置换价为多少?如利用此药在该栓模制备阴道栓8枚,内含主药每枚1.0g,需用可可豆脂多少?
解: (1)已知 W =1.5g、 G =3g、 M =3.7g,代入式(2-100)
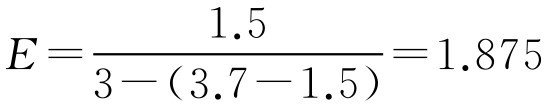
即1g可可豆脂需用1.875g药物来置换。
(2)已知 E =1.875、 W =1g、 G =3g,代入式(2-100)
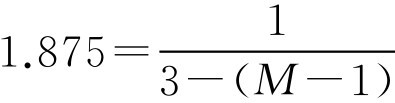
M =3.47(g)
即每颗栓剂中含基质的重量为3.47-1=2.47g,则做8枚阴道栓需用可可豆脂2.47×8=19.76g。
常用药物的可可豆脂置换价见表2-16。
表2-16 常用药物的可可豆脂置换价
2.容量法
容量法测定置换价实验方法和公式推导如下:将一定量熔融状的基质倒入一量筒中,读得体积为 V 1 ;再取一定质量药物加入到该基质中混匀后读得体积为 V 2 。


求得置换价后,可按下式计算按处方制备一定数量栓剂所用基质的量:

式中, V 为栓模能容基质的质量; Y 为欲制备栓剂的枚数; W 为每枚栓剂中药物的含量; E 为置换价; X 为制备这些栓剂需用的基质总质量。
【例2-54】 已由实验测得10g某基质体积为9ml,加入药物3g混匀后体积为11ml,空栓模可容2.2g该基质,要制备含药物0.5g的栓剂100枚,求栓剂的置换价及基质用量。
解: 根据式(2-101)、式(2-102)
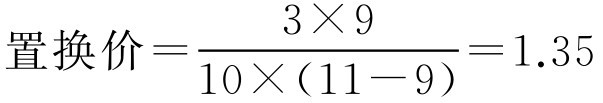
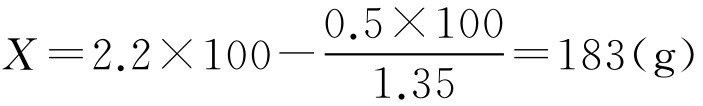
即该1g该基质需用1.35g药物来置换,按处方制备100枚栓剂需用基质183g。
1.氟氯烷烃类抛射剂的蒸气压与温度关系的有关计算
氟氯烷烃类抛射剂的蒸气压与温度的关系符合克劳修斯-克拉贝龙(Clausius-Clapeyron)方程

式中, p 为蒸气压,kPa;Δ H 为摩尔蒸发热,J·mol -1 ; R 为摩尔气体常数,8.314J·mol -1 ·K -1 ; T 为热力学温度; C 为常数。
通过式(2-103)可求得任何一个氟氯烷烃在任何一个温度时的蒸气压。
【例2-55】 已知F 12 的沸点为-29.8℃(即 T =243.4K), p =101.325kPa,21.1℃时( T =294.3K), p =585.0kPa,求其在54.4℃时的蒸气压。
解: 将数据代入式(2-103)得两个方程:
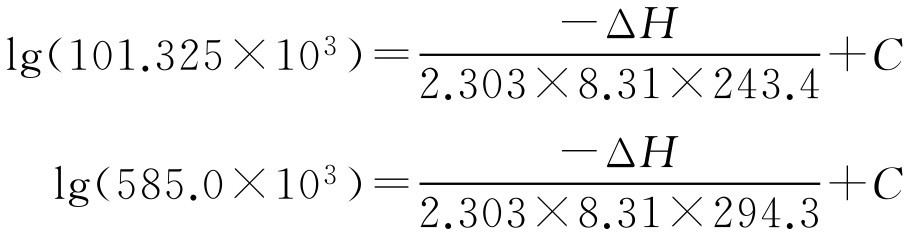
解方程得Δ H =20.47kJ·mol -1 、 C =6.400,即
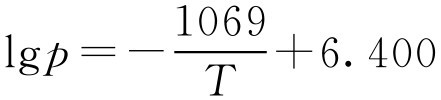
将 T =273.2+54.4=327.6K代入上式得 p =1370kPa。
即54.4℃时的蒸气压为1370kPa(实测值为1351kPa)。
2.混合抛射剂各组分的用量
“Raoult”定律指出:理想溶液中每个挥发成分的分压等于纯粹成分的蒸气压 p 0 乘以溶液中该成分的物质的量的分数,溶液的蒸气压等于各成分的蒸气分压之和,气雾剂中常用混合抛射剂的蒸气压可按这一定律计算:


式中,
p
a
为混合抛射剂中抛射剂a的蒸气分压;
p
b
为混合抛射剂中抛射剂b的蒸气分压;
N
a
为抛射剂a在混合抛射剂中的物质的量的分数;
N
b
为抛射剂b在混合抛射剂中的物质的量的分数,物质的量的分数的计算参阅第一章;
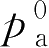 为纯抛射剂a的蒸气压;
为纯抛射剂a的蒸气压;
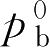 为纯抛射剂b的蒸气压。
为纯抛射剂b的蒸气压。
混合后的抛射剂的总压力为
p 总 = p a + p b (2-106)
常用抛射剂的物理性质见表2-17。
表2-17 常用抛射剂的物理性质
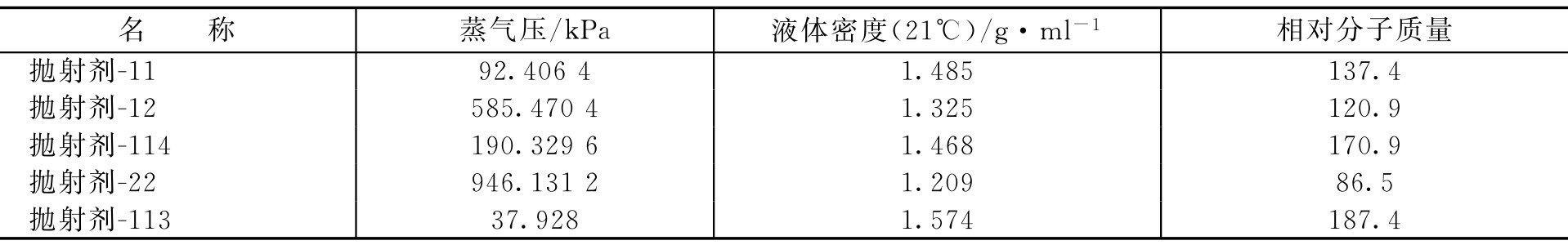
【例2-56】 选用抛射剂-12和抛射剂-114作混合抛射剂,欲获得275.84kPa压力(21℃表压)应各取多少?
解: 抛射剂-12和抛射剂-114的蒸气压(绝对压力)分别为:585.4704kPa和190.3296kPa,先换成相对压力。
绝对压力=表压(相对压力)+101.325
抛射剂-12的相对压力=585.4704-101.325=484.1454kPa
抛射剂-114的相对压力=190.3296-101.325=89.0046kPa
已知:
p
总
=275.84kPa,
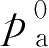 =484.1454kPa,
=484.1454kPa,
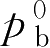 =89.0046kPa
=89.0046kPa
p a = N a ×484.1454 (1)
p b = N b ×89.0046 (2)
根据式(2-106)即得

即欲获得275.84kPa压力(21℃表压)的混合抛射剂,应取42.9ml抛射剂-12和61.7ml抛射剂-114。
多剂量给药的制剂,制成适宜的缓释(或控释)制剂,可减少给药次数及血药浓度波动,有利于提高用药顺应性及安全性。但要得到理想的血药浓度时间曲线,则应将缓控释制剂的释药速率与体内药物的吸收和处置动力学相结合,即根据需要的血药浓度和给药间隔进行剂量的设计。
1.控释制剂
当控释制剂以零级速率释放药物时,控释制剂的维持剂量
D
m
等于释放速率
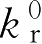 与维持时间
T
的乘积,即
与维持时间
T
的乘积,即


式中, k 表示消除速率常数; D b 为产生希望疗效的体内药量。
如按希望达到的稳态平均血药浓度(最佳血药浓度)计算,则


式中,
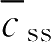 为稳态平均血药浓度;
V
为表观分布容积;
F
为吸收分数。
为稳态平均血药浓度;
V
为表观分布容积;
F
为吸收分数。
为了很快达到有效血药浓度,需要给予速释剂量 D f 。一般来说,服用速释部分剂量后,药物在体内迅速吸收达到有效血药浓度,即其剂量 D f 等于 D b 。
【例2-57】
已知某药最佳血药浓度
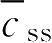 为4mg·L
-1
,
k
=0.2h
-1
,
F
=1,
V
=10L,释药时间为10h,试设计制剂中速释部分和控释部分药物用量。
为4mg·L
-1
,
k
=0.2h
-1
,
F
=1,
V
=10L,释药时间为10h,试设计制剂中速释部分和控释部分药物用量。
解: 根据式(2-110)、式(2-107)、式(2-109)得
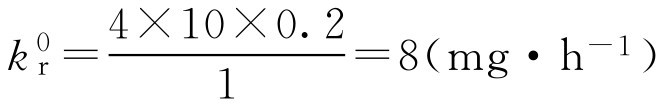
D m =8×10=80(mg)
D f = D b =4×10/1=40(mg)
由于实际上速释部分吸收达到峰浓度需要一定时间
t
max
,控释制剂也常以维持峰浓度
c
max
作为稳态平均血药浓度
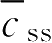 ,如果该药在体内符合一室模型,则
t
max
和
c
max
的求算公式为:
,如果该药在体内符合一室模型,则
t
max
和
c
max
的求算公式为:

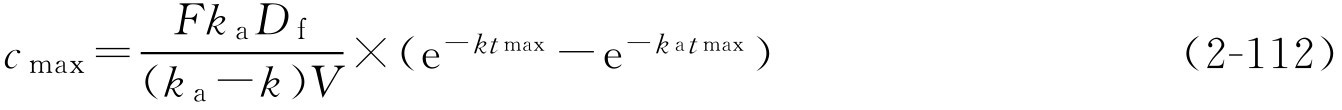
式中, k a 为吸收速率常数。
由于在速释部分释放药物的同时,缓释部分同时也释放药物,所以对上面的公式要进行校正。即速释剂量的公式校正为:

则控释制剂的总剂量 D tot 为速释剂量与缓释剂量之和,表示为:

【例2-58】 某药常规给药是每天4次,每次20mg,现研制每天给药2次的控释制剂,试计算制剂中药物含量。(已知 k =0.3h -1 , k a =2.0h -1 , V =10L, F =1)
解: 根据常规给药的剂量与给药间隔,计算重复给药的平均稳态血药浓度。(计算公式可参照药动学部分)
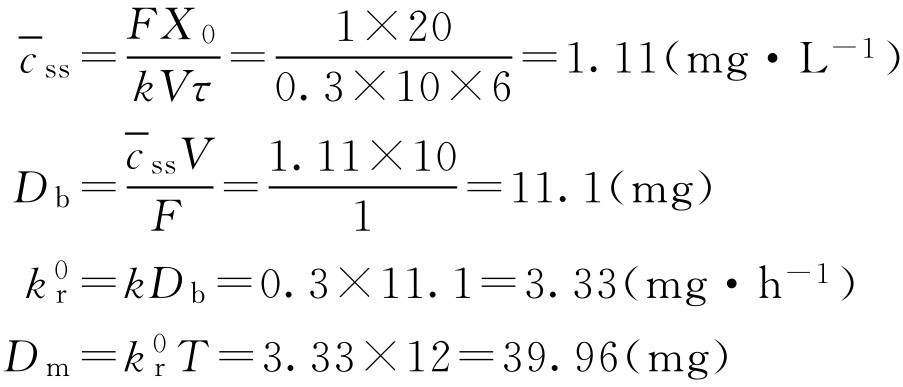
如该药在体内符合一室模型,产生希望血药浓度所需的速释部分剂量可用式(2-111)、式(2-112)计算。
将已知的条件和计算得的数据代入一室模型血药浓度公式中可解得 D f :
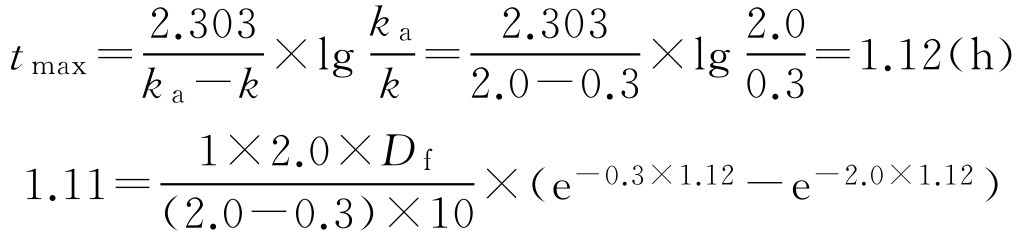
则 D f =15.5(mg)
如控释部分与速释部分同时释药,则代入式(2-113)和式(2-114)校正速释剂量:
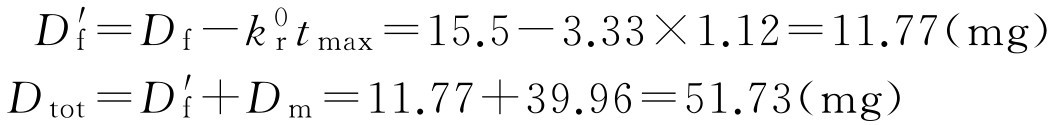
如果制剂设计控释部分是在速释部分释放后、血药浓度达峰值时释放的,则
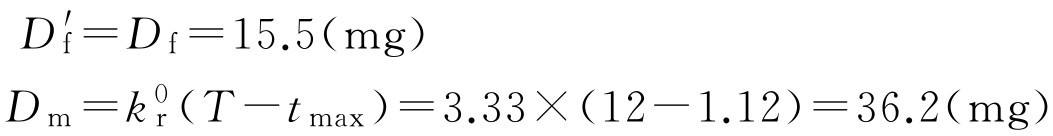
D tot =15.5+36.2=51.7(mg)
2.缓释制剂
缓释制剂中缓释部分剂量与药物的半衰期及希望维持治疗血药浓度的时间有关。表2-18为不同半衰期药物,缓释时间分别为6h、8h、12h的缓释与速释剂量的比值。
表2-18 不同半衰期药物的缓释速释剂量比
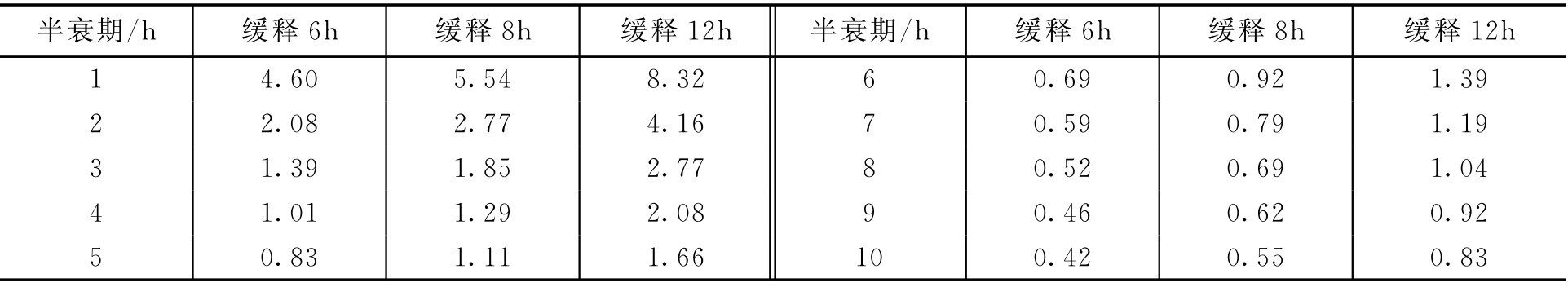
【例2-59】 某药的半衰期为4h,常用剂量为150mg,希望维持12h的治疗血药浓度,求其缓释剂量。
解: 查表得 D m / D f 为2.08,则缓释剂量 D m =150×2.08=312(mg)。
靶向制剂是指载体将药物通过局部给药或全身血液循环而选择性地浓集定位于靶组织、靶器官或细胞内结构的给药系统。利用水不溶性微粒易被单核巨噬细胞作为异物而吞噬,可将药物包封于不溶性微粒制剂(脂质体、微球与微囊、纳米球与纳米囊等)即可靶向于单核巨噬细胞丰富的肝、脾等组织,这种靶向作用称为被动靶向。如将这些微粒制剂进行表面修饰或选用特殊材料如磁性材料、温度敏感载体、pH敏感载体等还可达到主动或物理化学靶向。下面将这些微粒制剂的有关计算简述如下。
(一)包封率的有关计算
包封率又称包裹率或载药量,包封率的测定方法常有直接对制剂进行测定或将药物从制剂中分离出后进行测定。
1.质量包封率
是指包入制剂内的药物量与投料量的质量百分比,这样表示方法对脂质体、微球与微囊、纳米球与纳米囊均可用。
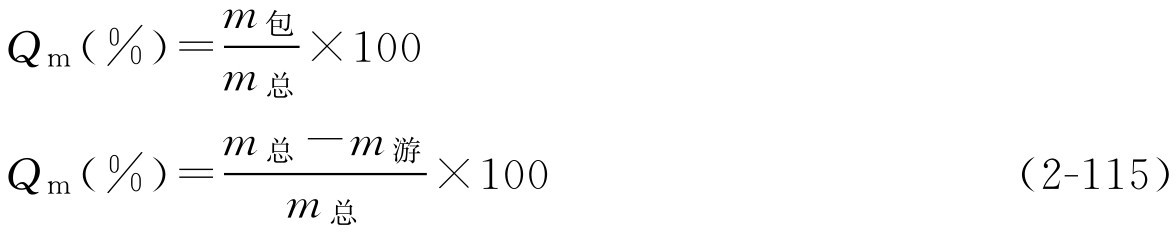
式中, m 总 、 m 包 和 m 游 分别表示投料量包封于脂质体等制剂的药量及未包入脂质体的药量。
2.体积包封率
是指制剂中某类粒子 V 类 与总体粒子 V 总 的体积百分比,可用于表示脂质体体积包封率,用下式表示:

式中, V 类 和 V 总 分别为脂质体制剂中某类粒子和总体粒子的体积。
Q v 的测定方法有凝胶过滤法和显微镜法等。
【例2-60】 已知用显微镜法测定某药物多相脂质体的体积包封率,测得结果如下:油球(O/W乳滴)和脂质体的比数为3.10/1000(50张照片的平均值),10个批号脂质体平均粒径为1.489 μ m,油球的平均粒径为3.61 μ m。试求该药物脂质体的体积包封率。
解: 用球形体积公式求得 V 脂 和 V 球 代入式(2-116):

即该脂质体制剂中脂质体的体积包封率为95.8%。
3.药脂百分比 E m
E m 表示一定质量的类脂所包封药物的质量百分比,用下式表示:
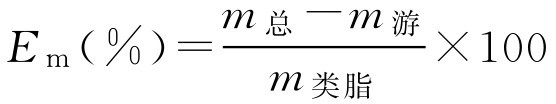

(二)靶向性评价的有关计算
药物制剂的靶向性可由以下三个参数衡量。
1.相对摄取率 r e

式中,AUC i 表示由浓度-时间曲线求得的某个器官或组织的药时曲线下面积,下标p和s分别表示药物制剂和药物溶液。 r e 值等于或小于1表示无靶向作用。
【例2-61】 分别给予大鼠某药物的溶液和微球制剂,测不同时刻肝、肾、肺中的药物浓度,并求得相应的AUC如下:
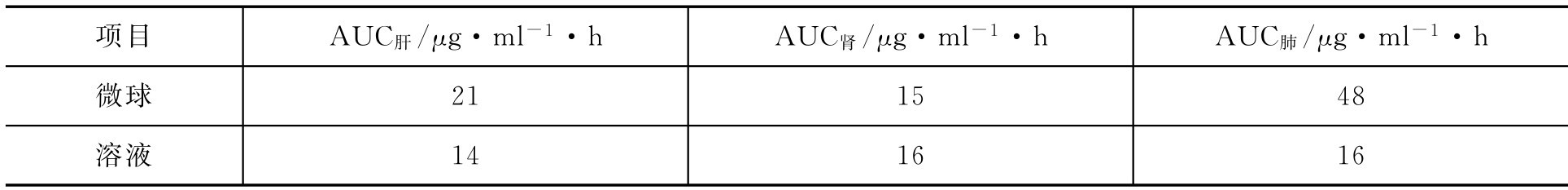
试判断该微球制剂对肝、肾、肺有无靶向性。
解: 根据式(2-118)
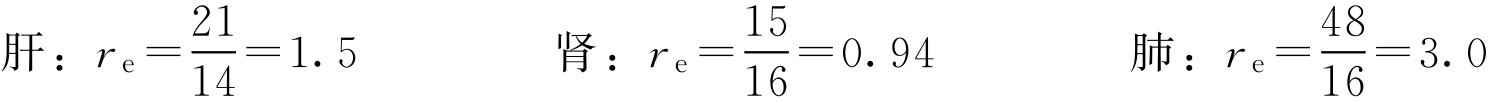
可见该微球制剂有较好的肺靶向作用,有一定的肝靶向性,无肾靶向作用。
2.靶向效率 t e
t e 表示靶向制剂或药物溶液对靶器官或靶组织的选择性。 t e 值大于1表示靶向制剂对靶器官或靶组织比某非靶器官有选择性, t e 值越大,选择性越大。

式中,AUC 靶 和AUC 非靶 分别表示靶器官和非靶器官的药物浓度-时间曲线下面积。
【例2-62】 数据见例2-61,试判断上述肺靶向微球制剂的靶向效率。
解: 根据式(2-119)
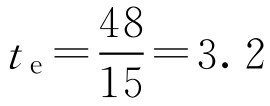
可见该肺靶向制剂对肺比肾的选择性高。
3.峰浓度比 C e
某药物靶向制剂和无靶向作用的药物溶液在每个组织或器官的峰浓度比 C e 表示靶向制剂可改变药物分布的效果,其值越大,表明改变分布的效果越明显。

式中, c max 为峰浓度,p和s分别表示药物制剂(靶向制剂)和药物溶液。